 电子发烧友App
电子发烧友App



目前已商业化的锂离子电池电极材料中的过渡金属存在溶解等交叉效应,严重影响着电池的循环性能。然而,当前关于交叉效应的研究大都基于氧化物正极的半电池,对氧化物正极和锂金属负极电池中交叉化学物质的影响知之甚少。
在本工作中,作者在具有高镍正极、锂金属负极和局部高浓度电解液(LHCE)的电池中探索了正极到负极和负极到正极交叉的影响。研究表明与高镍正极配对的锂金属负极的固体电解液界面(SEI)生长量是与锂金属配对的锂金属负极的三倍。同时,与锂金属配对的正极的容量衰减比与石墨配对的相同正极高2-3倍。其中FSI盐的分解和交叉被确定为这些变化的主要来源。
【内容详情】
为了区分一个电极对另一个电极的影响,作者用不同的电极组合组装了三组软包电池,包括具有高镍LiNi0.94Co0.06O2正极(NC9406或仅NC)和锂金属负极(Li)的电池(记作:NC|Li)、具有高镍正极和石墨负极(Gr)的电池(记作:NC|Gr)以及锂对称电池(记作:Li|Li)。
1. 电池和电极性能
如图1所示,具有LHCE、NC9406正极和锂金属负极的软包电池在200次循环后仍保持84%的容量。具有石墨负极的电池的容量保持率甚至更高,达到93%。与NC|Gr电池相比,NC|Li电池的容量衰减增加可归因于负极或正极与负极的相互作用(即交叉效应)。
通常考虑到锂金属负极的高反应性,人们直观地认为锂金属负极对NC|Li电池的容量衰减有很大贡献。然而,如图1c的对称电池数据所示,与直觉相反,锂金属负极通常在循环过程中表现出最小的阻抗增长。相反,一旦锂金属或电解液几乎耗尽,锂金属负极的反应性通常表现为快速失效。为了弄清本研究中锂金属负极在对称电池中的阻抗增长最小,但与NC9406配对时阻抗增长显着的现象,作者拆开循环的软包电池,并用循环电极的穿孔组以及新鲜的电解液和对电极装纽扣电池。通过这种方式,可以独立测量每个循环电极的容量和过电位。
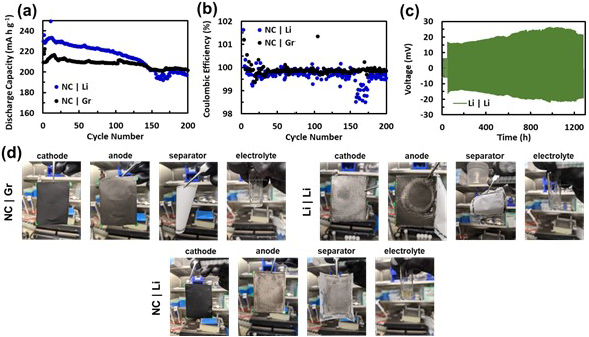
图1. 具有局部高浓度电解液的软包电池的循环性能和拆卸。(a) NC|Li和NC|Gr电池在循环过程中的比容量和(b)库仑效率;(c) Li|Li过循环的电压曲线;(d) 200次循环后循环电极、隔膜和电解液的图像。
有趣的是,与Li|Li的电池相比,具有NC|Li的纽扣电池最初确实具有降低的容量(图2b,2c)。这可能表明针对NC循环的锂金属负极确实比在对称电池中循环的阻抗增长更多。然而,这些纽扣电池的容量在5-10个周期内达到平衡,并且它们的电压曲线重叠良好,表明阻抗相似。尽管循环锂金属负极的早期润湿可能存在一些差异,这些重新组装的纽扣电池可比较的过电位和稳态容量表明NC|Li的锂金属负极对该电池的容量衰减没有贡献。因此,NC|Li电池相对于NC|Gr更大的衰减归因于正极。与循环正极组装的纽扣电池表明,具有NC|Li正极的纽扣电池在第一次循环中的容量明显低于具有NC|Gr正极的纽扣电池。与具有循环锂的纽扣电池不同,这种较低的容量也与增加的过电势相吻合,如图2c所示,较低的容量在整个循环过程中保持不变。此外,~18 mA h g-1的容量差异与NC|Li和NC|Gr软包电池的衰减差异相当合理,而NC|Li软包电池的最大容量比NC|Gr高约17 mAh g-1,最终容量比NC|Gr低约5 mA h g-1,变化量约为22 mA h g-1.
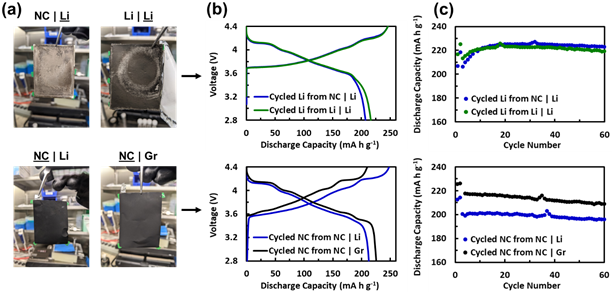
图2. 用新鲜电解液、新鲜对电极组装的纽扣电池和从拆卸的软包电池中回收的循环电极的循环性能。(a)显示用于重新组装电池的电极的图像;(b)电压曲线和(c)重新组装电池的循环数据。
2. 正极的体积和表面表征
如前所述,两个相同正极的容量衰减差异一定是由于不同负极的交叉效应造成的。要了解交叉对容量衰减的作用,有必要确定衰减的物理起源。就块状晶体结构而言,两个正极似乎都相对不受循环的影响。这在意料之中,因为高镍正极通常在几百个循环的过程中有最小的阳离子混合。虽然已知高镍正极会随着循环时间的延长而破裂,但这种现象通常在超过200次循环时也能观察到。此外,最近的研究表明,高稳定性电解液(如LHCE)通过抑制应力腐蚀开裂机制进一步减轻了开裂和微开裂,并且预计开裂导致该系统的容量衰减。
如果循环后的NC|Li的形态和晶体学变化很少,那么它的容量衰减与表面现象有关。为了探索这一点,作者用X射线光电子能谱(XPS,图3)分析了循环后的正极。正如预期的那样,每个正极都显示出来自溶剂分解的碳质物质以及来自FSI阴离子分解的含氮、硫和氟物质。有趣的是,对于与锂金属配对的正极,特别是氮和硫的正极,一些盐分解峰更为明显。虽然这可能表明NC|Li电池中的盐分解更大,但与盐分解和LiF形成相关的氟峰的强度反而降低了。同时,NC|Li的S 2p光谱不仅显示出硫的增加,而且还存在一些与元素硫和适度氧化的S-O相关的独特峰。相对于FSI的双氧合硫,这些硫物质被还原是出乎意料的。虽然与盐分解相关的氟、硫和氮光谱在NC|Li和NC|Gr之间存在显着差异,但碳和氧光谱相对没有变化。最显著的差异是在533 eV附近的O 1s的峰值,这归因于包括S-O、N-O和C-O在内的各种物种。虽然这些很难分成单独的峰,但具有更多硫和氮氧化物的样品应该具有更高强度的混合峰是合理的。
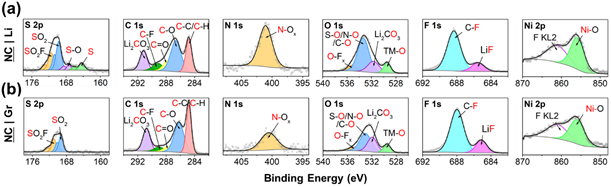
图3. 循环200次后循环NC9406正极的XPS数据:(a)正极与锂配对,(b)正极与石墨配对。
为了进一步探索循环正极的表面特性,作者还用飞行时间二次离子质谱(TOF-SIMS)对正极进行了分析。图4给出了NC|Li和NC|Gr正极的一些代表性分子片段的深度剖面,其中每个片段都归一化为其自身的最大强度。该数据中最引人注目的观察结果是以62Ni-为代表的镍信号的强度。该信号来自大块正极材料,该信号达到最大值的80%所需的时间/深度是衡量正极-电解液界面(CEI)厚度的指标。NC|Li电池的CEI约为40 nm,而NC|Gr电池的CEI约为140 nm。该结果与传统观点相反,经历了2-3倍容量衰减的正极的CEI薄了约3倍。为了证实这一结果的真实性,作者在同一样品的不同区域重复测量两次,并获得了相似的结果。在多次测量中看到的NC|Gr稍微“变薄”的CEI可归因于较短的溅射时间,这种情况下不允许62Ni-信号达到稳态。
综上所述,虽然NC|Li电池的CEI看起来比NC|Gr电池薄几倍,但是成分的变化似乎抵消了更薄CEI的好处。特别是与锂配对的正极(NC|Li)比与石墨配对的正极(NC|Gr)具有更大比例的硫和氮,而氟的比例更小。对于硫,这种成分差异部分表现为存在仅在NC|Li上发现的还原硫物质(S, S-O)。就有机CEI成分而言,样品之间的差异似乎很小。因此,盐分解物种似乎至少最初是交叉效应的主要驱动力。
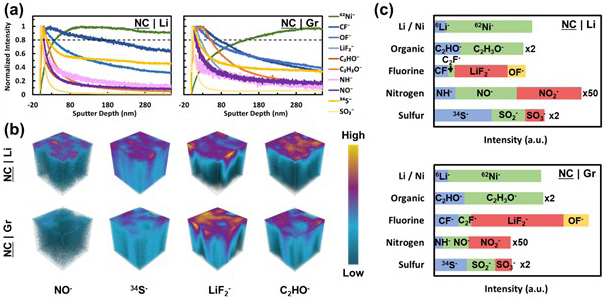
图4. 循环NC9406正极在200次循环后的TOF-SIMS数据。(a) 深度剖面显示几个特征分子片段的归一化强度;(b) 显示几个片段分布的3D图,样品面积100 x 100 μm,高度为350 nm;(c) 各种碎片的总综合强度,选择的碎片按比例放大以提高可见度。
3. 锂金属负极的体积和表面表征
按理说,如果锂金属负极影响正极,那么锂金属负极本身也应该受到影响。前面的结果已经表明,NC|Li和Li|Li电池的循环负极的阻抗增长在200次循环内是相当的。因此,有必要对锂金属负极SEI的物理特性进行表征。图5显示了循环锂金属负极的轴上和横截面SEM图像,揭示了SEI厚度的显着差异。尽管它们相似且有限的阻抗增长,NC|Li上的SEI比Li|Li上的SEI薄三倍。应该注意的是,本文中的“SEI”是指SEI组件和死锂的复杂框架,这是循环锂金属负极的典型特征。除了厚度之外,NC|Li的SEI形态也更致密且分辨率更好。这些图像是在放电(锂剥离)条件下拍摄的,因此应该更能表明SEI成分而不是锂金属形态。考虑到这一点,Li|Li样品的混浊及其缺乏明显的微观结构可能是由于较高比例的无定形有机物质所致。为了证实这一点并澄清SEI组成,作者再次使用XPS对Li|Li样品进行了表征。
为了避免由于NC|Li的锂金属负极出现充电问题对XPS测量准确性的影响,作者选择循环5次后的金属负极进行XPS测试,结果如图6所示。由于5个循环可能不足以让交叉物种达到稳定状态,因此该数据应被视为确定交叉效应的“最坏情况”。其中,NC|Li电池的氮和硫信号相对于Li|Li大幅减少了约40%。但是无论如何,氮和硫与氟的比例仍然在质量上符合预期:相对于氟,NC|Li电池上氮和硫的富集与NC|Li上的相反行为相关。

图5. (a) 循环锂金属负极在200次循环后的横截面和(b)轴上SEM图像,每个循环有两个放大倍数。
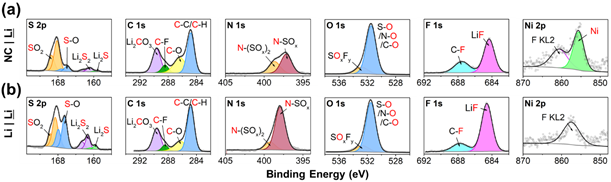
图6. 5次循环后循环锂金属负极的XPS数据:(a) 锂金属负极与正极配对,(b) 锂与锂配对。
在循环锂金属负极上硫和氮的键合环境中也可以看到一些明显的趋势。虽然Li|Li电池的硫含量通常高于NC|Li电池,但这种额外的硫主要处于与更多还原物质相关的较低结合能,例如Li2S、Li2S2和适度氧化的S-O。与循环正极相比,这很有趣,之前已经讨论过NC|Li电池具有更大比例的还原硫物质,例如S-O和元素硫。这可能与来自负极的还原物质的清除有关,这也可以解释相对于Li|Li,NC|Li电池上这些物质的含量较低。这可以描述为多硫化物穿梭的一种形式。在这种机制中,FSI中的一部分硫首先在锂金属负极上脱去氧,最终导致低阶硫化物,如Li2S和Li2S2。在电池放电期间,这些硫化物转化为可溶于DME溶剂的高级多硫化物,就像在锂-硫电池中一样。在NC|Li电池中,这些多硫化物穿过正极,在那里它们被氧化成元素硫和硫氧化物。与锂-硫电池不同,正极电压永远不会低到足以将硫还原成多硫化锂,因此“多硫化物穿梭”是单向且不可逆的。石墨,没有金属锂容易还原SO2,形成这些硫化锂的速度较慢,因此循环NC|Gr电池没有测到元素硫。
NC|Li和Li|Li电池上氮的键合环境也不同(图6)。NC|Li电池上的平均结合能和氧化态低于Li|Li。在N 1s中看到的这种更大比例的N-(SOx)键与在S 2p中看到的SO2相对于S-O的更高浓度相匹配。显然,盐在NC|Li上的分解不太彻底,或者那些分解程度更高的分子通过交叉去除。在有机物种方面,两个锂金属负极样品非常相似,无法分离各种氧物种。虽然NC|Li的碳谱似乎确实相对于Li|Li有适度的CO-O增加,但其碳谱在其他方面是相同的。C-O键的增加可归因于多种机制,例如由富氟SEI驱动的溶剂分解差异,或来自正极的氧化溶剂种类的交叉。
因此,关于锂金属负极的主要结论反映了循环正极的结论。NC|Li的SEI/死锂层厚度是Li|Li上厚度的三分之一,其中SEI厚度是稳定性的直接衡量标准,因为它代表了活性锂的损失。NC|Li相对于氟富含硫和氮,而NC|Li相对于硫和氮富含氟(分别与NC|Gr和Li|Li相比)。这可以解释为FSI中的氟在硫和氮穿梭到正极之前被锂金属负极优先清除,反之亦然。此外,作者还发现了单向多硫化物穿梭的证据,这进一步耗尽了负极中的硫并使其在正极上富集。作者在循环锂金属负极(和循环石墨)上发现了过渡金属沉积物,尽管它们似乎不太可能促成SEI厚度减少3倍。与正极一样,溶剂交叉效应对锂金属负极的影响很小。
4. 电解液分析和交叉机制
为了进一步研究哪些物种可能参与了交叉,作者从循环电池中回收电解液并使用傅里叶变换红外光谱(FTIR)和核磁共振光谱(NMR)对其进行表征,结果表明电解液的整体组成几乎保持不变。
最终,FSI盐阴离子被提议作为该系统中交叉效应的主要驱动因素,甚至超出了其作为多硫化物穿梭的硫源的作用。与其他机制相比,基于FSI的交叉解释了更多观察到的表面征,并且鉴于已知盐分解在浓缩电解液中占主导地位,因此是直观的。该机制可以描述如下:在锂金属负极上,FSI分解的第一步是除氟。在锂对称电池中,产生的脱氟FSI在两个电极上形成,并继续在电解液中积聚。最终,脱氟的FSI开始与锂金属反应,将其氮和硫添加到锂SEI中。同时,在具有锂金属负极和高镍正极的电池中,脱氟FSI无法在电解液中积聚。相反,它会越过正极,在那里脱氟FSI较低的氧化稳定性导致其快速消耗。这种脱氟FSI与正极的交叉减少了锂金属SEI中的硫和氮,并允许FSI连续脱氟,从而增加氟含量。正极受到相反的影响,硫和氮增加,氟含量减少。正极表面成分的这些变化通过带有石墨负极的电池得到证实,与锂金属负极相比,石墨负极向正极输送的脱氟FSI物质更少。
鉴于氟作为SEI保护成分的已知状态,交叉对氟含量的影响可以解释锂金属负极与高镍正极配对时的出色稳定性。同样,氟含量的降低可以解释正极在同一系统中稳定性较差的原因。硫和氮的含量也可能起作用,尽管这不能与氟的影响分开。虽然基于FSI的交叉在所研究的系统中似乎是有害的,但必须考虑到本研究中使用了过量的锂和电解液,在锂稀薄的系统中,跨接实际上可以通过保护锂金属负极来延长循环寿命,而锂金属负极通常是锂金属电池中的限制电极。这项工作还进一步证明,无论是通过添加剂还是更重氟化的盐,SEI和CEI中的氟含量越高,可以实现循环寿命的提高。但是,添加剂或盐改性对溶解度、热稳定性等仍有严格的要求。最后,必须注意的是,在具有多重交叉效应的系统中,很难完全分得清因果关系。因此,该系统还有很大的进一步研究和优化空间。
审核编辑:汤梓红












 工商网监
工商网监
评论