纸电池:能源储存新兵
A4纸大的动态电子报纸、自动计价的超市、会“说话”的包装盒……几年后,一张0. 5毫米厚的纸电池将创
2010-02-05 09:10:56 1066
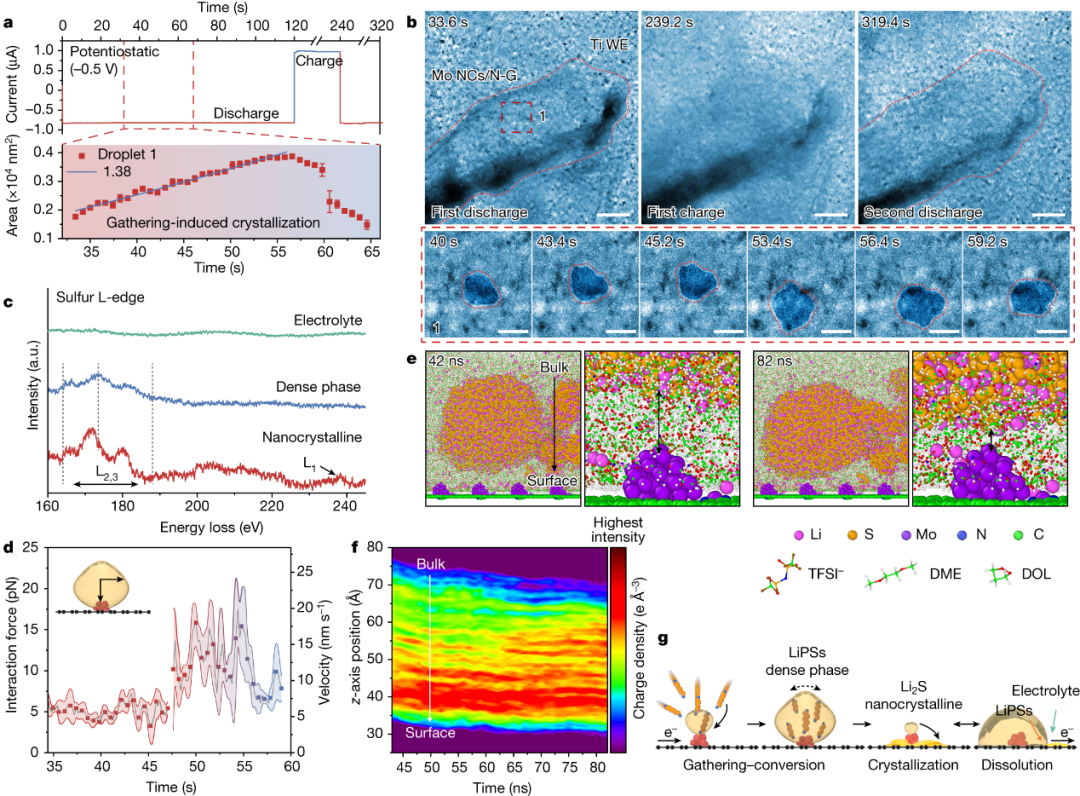
1066 一种解决传统电解质不稳定的方案是使用熔盐电解质,有研究表明,Li-O2电池可以在熔融硝酸盐(LiNO3/KNO3)中循环,并具有可逆双电子氧还原,仅具有0.1 V的电压滞后。
2022-08-22 10:42:242319 浅谈公共机房样机制作及日常维护随着电脑和网络的应用日益普及, 电脑在日常生活中扮演着越来越重要的角色,而学校的教育,无论是高校还是中小学,公共机房都是一个不可缺少的教学实践基础硬件设备。而如何高效
2009-10-10 15:04:47
),都是在把电网的电能传输到汽车的电池上进行储存,因为电池只能储存直流电,所以一般交流电是不能直接到电池的,一般都得通过车载充电机进行(OBC)AC/DC的转换;
2021-04-19 17:19:55
使用 ADS7142 SAR ADC 和 TLV521 运算放大器实现低功耗系统监控单通道负载开关,可连接/断开系统负载支持锂亚硫酰氯 (LiSOCl2) 和锂二氧化锰 (LiMnO2) 电池系统运行状况监控待机功耗和电池监控待机功耗均低至 1uA
2018-12-28 15:11:47
楼主翻阅新闻的时候发现电池续航能力好像又有新的进展了,而且可储存锂电池五倍电量。我们一起看看是什么样的技术能做到如此吧。 《自然》上发表了一项研究,描述了一种“锂氧电池”的原型,其基于超氧化锂
2016-01-29 14:38:28
锂二氧化锰电池的反应机理不同于一般电池,在非水有机溶剂中,负极锂溶解下的锂离子通过电解质迁移进入到MnO2的晶格中,生成MnO2(Li+)。Mn由+4价还原为+3价,其晶体结构不发生变化。
2020-03-10 09:00:32
以下关于锂电池各蓄电池的标准内容是我司工程师花了一两小时整理出来的,优耐检测带大家来了解一下:1高空模拟 锂原电池和蓄电池在运输中的安全要求 GB 21966-2008 IEC 62281:2016
2020-12-25 15:29:13
能技术完全不同,击败锂离子电池的潜力极大。这种电池的用金属锂做负极,在正极一端直接与空气中的氧气反应。由于反应物之一是空气,理论上讲,该电池储存同样能量所需材料仅为其他电池的一半,其重量也可减半。这一
2018-10-09 10:28:23
还缺乏对何种电极材料能够促进电池内部电化学反应发生的理解。Shao-Horn和其团队成员在4月1日出版的《电化学与固态快报》上报道了其研究成果,在锂-空气电池中使用金或铂金电极作为催化剂具有比单一碳
2016-01-13 16:04:23
锂铁电池的内部结构如图1所示。左边是橄榄石结构的LiFePO4作为电池的正极,由铝箔与电池正极连接,中间是聚合物的隔膜,它把正极与负极隔开,但锂离子Li+可以通过而电子 e-不能通过,右边是由碳(石墨)组成的电池负极,由铜箔与电池的负极连接。
2019-09-30 09:10:42
锂锰电池在生产过程中使用了低沸点的有机物溶剂,其中有一种叫乙二醇二甲醚(DME)的物质,其闪点温度较低。在充电过程中,如果电池密封不好,电池发热造成该物质的挥发,遇到电火花将有可能发生燃烧,产生危险。
2019-11-06 09:10:46
Android系统固件更新机制设计说明文档V1.1xxx2014-9-14修改历史记录内容编制\日期审核\日期批准\日期 V1.0建立初稿Xxx2014-9-14 V1.1 增加配图,统一英文单词大小写Android启动过程错误修正,红色字体部...
2021-12-20 08:08:24
一种新颖的环路内去块效应滤波器设计,设计中采用5阶流水线的去块效应模块,利用混合滤波顺序与打乱的存储更新机制的方法提高了流水线畅顺性,滤波一个16×16大小的宏块仅需要198个时钟周期。
2021-04-12 06:35:37
LED光源怕硫,这是因为含硫的气体会通过其多孔性结构的硅胶或支架缝隙,与光源镀银层发生硫化反应。LED光源出现硫化反应后,产品功能区会黑化,光通量会逐渐下降,色温出现明显漂移;硫化后的硫化银随温度
2014-09-12 09:52:15
一般说明
PL5353A产品是锂硫离子/聚合物电池保护的高集成解决方案。
PL5353A包含先进的功率MOSFET,高精度电压检测电路和延迟电路。
PL5353A被放入超小型SOT23-5封装中
2023-11-07 10:23:19
PSRAM就是伪SRAM,内部的内存颗粒跟SDRAM的颗粒比较相似,但外部的接口跟SDRAM不同,不需要SDRAM那样复杂的控制器和刷新机制,PSRAM的接口跟SRAM的接口是一样的。psram内部
2020-12-28 06:43:38
1.请教一下,像TC275HSM 能支持SecOC中的密钥刷新机制吗?即SecOC中的密钥生成节点生成新的密钥后,是如何下发给ECU的HSM中?2. 二代HSM TC3xx 的HSM 集成
2024-02-21 06:07:23
范围-55ºC至+85ºC锂金属含量约 2.5克U.L. 组件识别,MH 121933.6 V伯亚硫酰氯锂(Li-SOCl2)长期存储和/或使用后,电压恢复很快高能量密度低自放电率梭芯结构密封玻璃到
2020-11-18 11:28:42
要的。
幸运的是,Oculus和Crytek一起在VR移动机制的探索上花费了不少时间,现在他们也将成果开放给大家。
在博客中,作者强调开发者应当选取正确的移动机制,或者一系列移动方式的结合,来
2017-08-31 09:17:12
深圳市尊信电子技术有限公司 唐名江 *** WX:tmj8213XB3306A产品是锂/聚合物电池保护的高集成度解决方案。XB3306A包含先进的功率MOSFET,高精度电压检测电路和延迟电路
2021-04-07 14:03:02
的TTL电平定时器和终端启用(TTE)输入终止。设备信息功能框图功能描述bqTINY™ 支持适用于单电池的精密锂离子锂极化充电系统。图2显示了典型的电荷分布、应用电路,图5显示了操作流程图。温度鉴定注意
2020-09-21 18:17:03
LED光源怕硫,这是因为含硫的气体会通过其多孔性结构的硅胶或支架缝隙,与光源镀银层发生硫化反应。LED光源出现硫化反应后,产品功能区会黑化,光通量会逐渐下降,色温出现明显漂移;硫化后的硫化银随温度
2019-07-09 10:26:51
` 检测煤炭含硫仪器 酒泉煤炭测硫仪,检测煤炭含硫仪器 酒泉煤炭测硫仪由【鹤壁化验煤炭含硫仪器】提供的检测煤炭含硫仪器 酒泉煤炭测硫仪测硫机:***化验机0392-2121168煤炭快速测硫仪
2019-03-21 20:39:10
锂铁电池是近几年才兴起的新一代干电池,其具有容量大、体积小、重量轻、大电流放电、低温性能优异的特点。相较于碱性电池而言,锂铁电池不易漏液,因为电解液完全吸附在隔膜上,电池内部不存在流动性液体,不会
2018-11-27 13:21:49
%。自放电率低(年自放电率≤1%)、储存寿命长达10年以上。 以1#(尺寸代码D)镍镉电池与1#锂-亚硫酰氯电池的比能量作一个比较:1#镍镉电池的额定电压为1 2V,容量为5000mAh;1#锂-亚硫
2014-12-03 18:05:58
(ZnCl2)、锌二氧化锰(ZnMnO2,即碱性电池)。 还有一系列的锂电池:锂-二硫化铁(LiFeS2)、锂-二氧化锰(LiMnO2)、亚硫酰氯锂(LiSOCl2)、氟化碳锂聚合物(LiCFX)、锂
2014-08-18 09:33:17
~+70°C (特定产品可低至-60°C)。化学反应式阳极:Li → Li+ + e-阴极:2SO2 +e → S2O4整体:2Li + 2SO2 → Li2S2O4用途虽然锂二氧化硫电池是以军事应用为
2014-08-18 10:30:58
在即使是恶劣条件下的排放也十分有限的特性可说是非常优异,可惜的是其电解液具有毒性,并会与水产生反应。此外锂亚硫酸氯电池具备高能量比、重量轻,但内部阻抗非常高,因此只能以有限的短路电流低速放电;还有一个
2014-08-18 10:20:42
二极管的储存电荷
2021-03-03 07:10:48
储能电池模块(锂原材料)篇 行业概述随着城市建设中能源危机和环境污染问题,2021年结合我国实施执行碳中和的远大目标和大环境下,实现风光互补发电系统多行业应用推行,具备良好超前的发展前景空间
2022-03-11 15:59:46
一次锂电池(锂亚硫酰氯电池,锂硫酰氯电池)的主要市场用途: 1) 检测仪表: 热量计;自动仪表读数器(AMR)---如水表、气表或电表等;汽车试验场检测仪;地震测量仪;石油钻探检测仪器;资料记录器
2015-01-26 10:13:21
一次锂电池(锂亚硫酰氯电池,锂硫酰氯电池)的主要市场用途: 1) 检测仪表: 热量计;自动仪表读数器(AMR)---如水表、气表或电表等;汽车试验场检测仪;地震测量仪;石油钻探检测仪器;资料记录器
2015-01-26 10:23:56
一次锂电池(锂亚硫酰氯电池,锂硫酰氯电池)的主要市场用途: 1) 检测仪表: 热量计;自动仪表读数器(AMR)---如水表、气表或电表等;汽车试验场检测仪;地震测量仪;石油钻探检测仪器;资料记录器
2015-01-26 10:28:54
一次锂电池(锂亚硫酰氯电池,锂硫酰氯电池)的主要市场用途: 1) 检测仪表: 热量计;自动仪表读数器(AMR)---如水表、气表或电表等;汽车试验场检测仪;地震测量仪;石油钻探检测仪器;资料记录器
2015-01-26 10:30:38
一次锂电池(锂亚硫酰氯电池,锂硫酰氯电池)的主要市场用途: 1) 检测仪表: 热量计;自动仪表读数器(AMR)---如水表、气表或电表等;汽车试验场检测仪;地震测量仪;石油钻探检测仪器;资料记录器
2015-01-26 10:33:46
一次锂电池(锂亚硫酰氯电池,锂硫酰氯电池)的主要市场用途: 1) 检测仪表: 热量计;自动仪表读数器(AMR)---如水表、气表或电表等;汽车试验场检测仪;地震测量仪;石油钻探检测仪器;资料记录器
2015-02-02 13:01:23
,积极探索管理新机制。街道纳管辖区45家电动自行车生产、销售、维修单位,并化繁为简,采取“疏堵结合、以疏为主、***搭台、企业运作、智慧管理”模式,以电动车保有量为413台的沙塘布社区为实施试点,计划建设6个
2018-08-29 14:51:42
颗粒,在正极侧接触,这是难度非常大的。从大家预期的优点上,如果使用了金属锂,现在容易燃烧和爆炸的液态电解质,另外使用寿命等等都会延长,模块配置等都是大家期望的,包括金属锂、锂硫和锂空气电池,这些路线在
2017-01-17 09:37:14
和可再生能源过渡的立法,这种短缺将变得更加严重。因此,市场迫切需要新的电池技术来解决锂短缺问题。锂在元素周期表上的地位几乎奠定了其能量密度之王的地位。一方面,锂在可再生能量储存方面使用寿命成本很低,优势
2021-04-06 15:03:35
为了研制在电性能、安全性和成本价格等三方面均能较好地满足电动汽车需求的锂离子电池,选择了在氧化钴锂中掺杂氧化镍锰钴锂三元材料的方法,研制了新的50Ah动力型锂离子电池。通过对研制电池进行电性能
2011-03-04 14:30:54
,但是将锂空气电池用于商业用途至少还需要10年的时间。 锂空气电池的基本化学原理十分简单。放电时,从负极出发的锂离子在正极与空气中的氧气反应,产生一种叫过氧化锂的固体产物,填充于碳电极的孔隙中。充电
2016-01-11 16:15:06
浅谈新能源汽车电源之锂硫电池利与弊 一直以来,新能源汽车用动力电池能量密度小和造价高的问题一直困扰着国内外新能源汽车制造企业,为了破解这一难题,近来动力电池生产企业纷纷开始研发
2018-07-13 07:54:40
求帮助啊。我现在一脸懵逼。不太懂刷新机制,想做个电子书但是无奈编程太渣。求大牛指点啊
2016-05-11 09:06:10
检测硫的设备—化验硫含量的仪器 检测硫的设备—化验硫含量的仪器【全自动测硫仪】英特仪器详询:138.3923.4904 煤炭测硫仪,高精度全自动定硫仪,微机快速定硫仪,生物质含硫量分析仪,化验石油焦
2020-12-28 11:05:45
`检测萤石全硫总硫仪器哪些设备准确 检测萤石全硫总硫仪器哪些设备准确 英特仪器萤石含硫量测定仪,测试萤石总硫的仪器,化验萤石全硫含量的设备,测量萤石硫含量的仪器,萤石含硫量分析仪详询
2021-04-13 09:00:45
本文提出了一种具有较高稳定性和安全性、基于bootloader的嵌入式软件自动更新机制。该更新机制同时保存了3个文件,需要较多的Flash存储空间,但同时降低了维护成本。
2021-04-27 06:33:59
测硫仪部件结构及功能 定硫仪主要包括高温裂解炉、送样机构、电解池和搅拌器、空气净化系统、控制系统等。
2019-10-23 09:01:03
电力能源的使用,可用的能量的量很小,往往是不可靠的。传感器节点通常需要某种方式储存能量暂时使它准备阅读时必须采取或信息无线发送。选择之一是提供一种小型可充电电池或储能电容器。然而,这些存储机制有其自身
2016-03-02 16:46:48
磷酸锂铁电池广泛应用于电动车、油电混合车、电动自行车、电动工具机、太阳能LED路灯等。
2019-10-22 09:01:28
锂空气电池是一种用锂作阳极,以空气中的氧气作为阴极反应物的电池。 放电过程:阳极的锂释放电子后成为锂阳离子(Li+),Li+穿过电解质材料,在阴极与氧气、以及从外电路流过来的电子结合生成氧化锂
2016-01-11 16:27:12
结构紧凑的锂(Li)电池充电器设计方案
2009-03-26 22:02:01
,理论上正极的容量密度是无限的,可加大容量。另外,如果负极使用金属锂,理论容量会比锂离子充电电池提高一位数。但是,为什么锂-空气电池至今都未普及?原因是它存在致命缺陷,即固体反应生成物氢氧化锂(LiOH
2016-01-12 10:51:49
磷酸铁锂电池储能系统 磷酸铁锂电池具有工作电压高、能量密度大、循环寿命长、绿色环保等一系列独特优点。并且支持无级扩展。组成储能系统后可进行大规模电能储存。 磷酸铁锂电池储能系统由磷酸铁锂电池
2015-10-20 16:13:21
信号和槽作为Qt的和新机制,在Qt编程中有着非常广泛的应用。 事实上,我们在Qt开发中,要想妥善的处理各种事件,根本就离不开信号与槽。信号是事件,信号通过信号处理程序来响应。当一个信号被发射时,相应的信号处理程序就会被调用,在处理程序中编写代码来使控件响应事件。
2020-11-20 08:03:11
钠硫电池是一种用于非移动设备如栅极储能的熔融金属电池。钠硫电池由钠和硫组成,与其他电池相比,具有非常高的能量密度和非常高的充放电效率。钠硫电池是由一层固体电解质膜构成的电池。钠硫电池具有很高的能量
2022-04-28 10:53:27
是使用二氧化锰为正极材料、金属锂或其合金金属为负极材料、使用非水电解质溶液的电池。放电反应:Li+MnO2=LiMnO2(2)锂离子电池:锂离子电池一般是使用锂合金金属氧化物为正极材料、石墨为负极材料、使用非
2018-03-31 14:19:48
锂电池包的新机遇,回收再利用!我们首先想到的是家用废电池和废铅蓄电池。而不被人所熟知的,锂动力电池作为新兴的储能形式开始被人们所接受。 虽然锂电池包结构复杂,规格不一,回收再利用难度比较大。 但
2018-08-16 09:25:07
【锂知道】锂电池基本原理解析:充电及放电机制电池充电最重要的就是这三步:第一步:判断电压
2021-09-15 06:47:08
滤布的孔隙从净气室排出,脉冲电磁阀有规律地控制压缩空气滤布表面的粉尘,粉尘脱离滤布坠入料仓。
五、烧结:高温烧结工段主要反应原理为,磷酸铁、碳酸锂和碳源在以氮气为保护气的条件下在电加热辊道窑中反应成磷酸铁锂
2023-09-12 13:22:46
很多人会误以为锂离子电池就是锂电池,实际上两者是有区别的。那么锂离子电池和锂电池的区别在哪里呢? 锂电池的正极材料是二氧化锰或亚硫酰氯,负极是锂。举例来讲,以前照相机里用的扣式电池就属于锂电池
2015-12-28 15:10:38
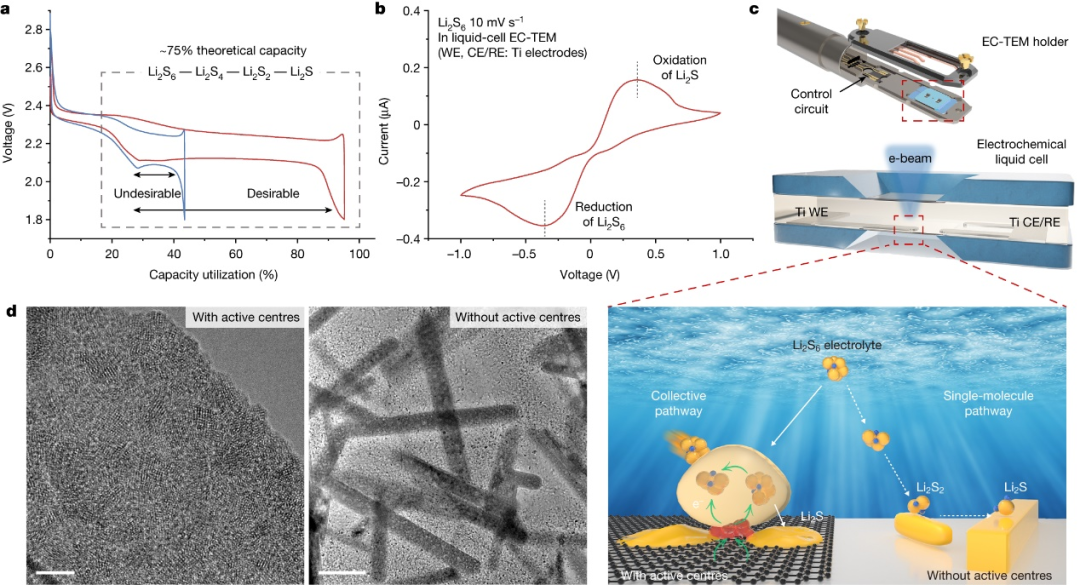
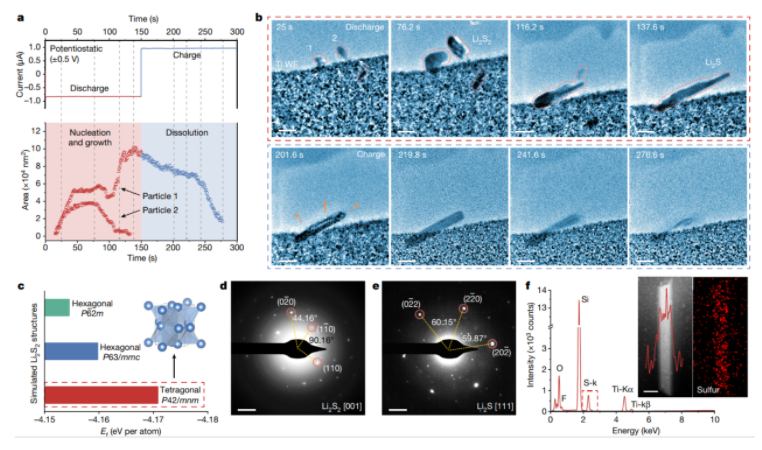
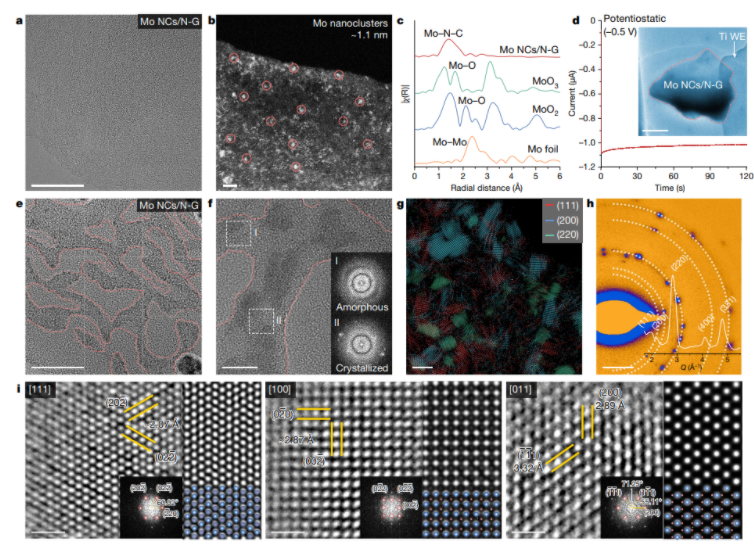
|Li电池上这些物质的含量较低。这可以描述为多硫化物穿梭的一种形式。在这种机制中,FSI中的一部分硫首先在锂金属负极上脱去氧,最终导致低阶硫化物,如Li2S和Li2S2。在电池放电期间,这些硫化物转化
2022-08-30 08:15:15
新型电池、新型能源不停的进步发展,作为老前辈的锂电池也不甘落后,最近日本又研发出锂离子电池的最新正极材料-掺锰铌酸锂,据说能量密度有望达6倍,我们快来看看这种正极材料到底是什么,为什么这么厉害吧
2016-01-19 14:06:07
的市场需求。在过去几十年中,研究人员提出了多种新型负极材料,这些材料通常表现出理想的电势范围、更高的容量、优异的倍率性能以及长的循环寿命等优势,但具有初始活性锂损失较大(ALL)这一不足。因此,在全电池
2021-04-20 16:15:15
钴酸锂、镍酸锂和锰酸锂化合物是近年来锂离子蓄电池最具吸引力的三种正极材料。从比能量、环境污染和价格方面看,锰酸锂化合物最具前景。重点阐述了尖晶石型锰酸锂化合物的制备方法,诸如:固相反应法
2011-03-10 12:46:42
提出了一种基于嵌入式系统的远程程序更新机制,通过一个具体的嵌入式远程数字监控系统设计方案,分析了该机制的系统结构、实现原理和实现流程,实际的应用测试表明,所
2009-08-26 11:47:41 14
14 ADO_NET数据集更新机制及并发控制策略:本文分析了8I5J (?> 中的更新机制,论述了三种不同的更新逻辑的产生方式及各自特点,提出了并发控制的一些解决方法,及更新逻辑中其他一
2010-01-01 18:48:3112 嵌入式系统自更新机制的设计与应用
随着嵌入式系统的发展和广泛应用,必不可少的维护工作变得日益繁重。如移动电话在用户使用过程中,部
2009-03-29 15:08:02762 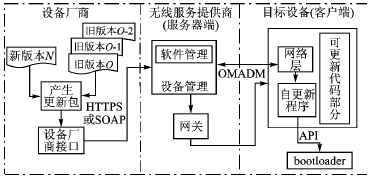
电池储存期有多久?
就理论上讲,电池储存时总有能量损失。电池本身固有的电化学结构决定了电池容量不可避免地要损失
2009-10-20 13:41:262103 电池储存应该怎样存放
根据 IEC 标准规定,电池应在温度为 20+-5O ° C ,湿度为( 65-+20 ) % 的条件下储存。一般而言,电池储存温度越高,容量的剩
2009-10-20 13:42:404337 电池能储存多久?
就理论上讲,电池储存时总有能量损失。电池本身固有的电化学结构决定了电池容量不可避免地要损失,主要是由于自放电造成的。通常自放电大
2009-10-28 14:21:56657 Tadiran 电池的储存与运输常识
一般性预防措施
2009-10-30 13:07:001569 电池为什么要半电储存?
2009-10-30 14:26:541452 电池能储存多久?
就理论上讲,电池储存时总有能量损失。电池本身固有的电化学结构决定了电池容量不可避免
2009-11-13 13:36:403939 氢氧电池反应原理图
2009-12-18 09:02:58949 文中所提出的新机制采用敲击键盘选择口令图片,代替了以往由鼠标点击的选择方式,其优点在于可有效地防止肩窥攻击,并能抵御交叉攻击,增加了口令机制的安全性。实验证明,该机制具有
2012-02-13 16:24:2725 全国人大代表、中国科协副主席、中国工程院院士邓中翰院士表示,将在两会期间提交三个建议,其中包括“进一步深化科技体制改革、完善自主创新机制”的建议。
2012-03-03 14:56:23642 为满足航天应用中数据传输与存储中高可靠以及低功耗的要求,实现了一种自适应刷新机制的同步动态随机存储(Synchronous Dynamic Random Access MemorySDRAM)控制器
2018-04-03 16:00:440 在美国加利福尼亚州旧金山举行的IEEE第64届国际电子设备会议上,三菱电机公司和东京大学提出了提高SiC功率半导体可靠性的新机制。 这个新机制是通过确认栅极氧化物和SiC之间的界面下的硫捕获器件电流
2019-05-04 12:45:00631 全钒液流电池是将具有不同价态的钒离子溶液分别作为正极和负极的活性物质,分别储存在各自的电解液储罐中。在对电池进行充、放电实验时,电解液通过泵的作用,由外部贮液罐分别循环流经电池的正极室和负极室,并在电极表面发生氧化和还原反应,实现对电池的充放电。
2019-12-02 11:26:1110565 对分层式三维室内地图分类方法及更新机制说明。
2021-03-17 15:52:510 电子发烧友网为你提供化敌为友,二极管的储存电荷资料下载的电子资料下载,更有其他相关的电路图、源代码、课件教程、中文资料、英文资料、参考设计、用户指南、解决方案等资料,希望可以帮助到广大的电子工程师们。
2021-04-06 08:43:564 电容器充电的过程,开关s闭合以后,A极板就通过导线、开关与电源E的负极连接,B极板通过导线与电源正极连接。由于电池正极聚集大量正电荷,就将导线ab中的负电荷吸到a端来,b端就剩下正电荷,在导线b端正电荷作用下,又将B极板中负电荷吸出来,B极板就只剩下正电荷。
2021-06-02 15:36:01918 电池的自放电为什么重要?电池需要多长时间才会自放电? 电池的自放电是指电池在不使用时,由于内部自身的化学反应而自发地失去电荷。这个现象对于电池的使用非常重要,因为它直接影响了电池的储存能力和使用寿命
2023-11-10 15:01:10523 锂电池组如何储存呢? 锂电池组的储存是非常重要的,它可以确保电池的长期稳定性和延长寿命。储存不当可能会导致电池组容量下降、功率衰减,甚至引发安全问题。因此,在储存锂电池组之前,有一些关键的因素需要
2024-01-10 10:30:29314 高表面积的电极(通常是活性炭)之间的电解质进行电荷的存储。当电力加到超级电容器时,正负电荷分开存储在两个电极上,不涉及化学反应。当电流从超级电容器中流过时,这些储存的正负电荷被释放。 1.2 电池的工作原理 电池是通过化学反应
2024-02-04 10:12:31447 电容器接在电路上的作用是什么?会使正负电荷聚集吗?电压与电容器并联会怎样 电容器是一种用于储存电能的器件,它在电路中具有多种重要功能。首先,电容器能够储存电荷。当电容器正常工作时,电荷会在电容器
2024-02-06 11:24:00380 坚果派由坚果创建,团队拥有 12 个华为 HDE,以及若干其他领域的三十余位万粉博主运营。团队成员聚集在北京,上海,南京,深圳,广州,宁夏等地,欢迎合作。 首先铺垫两个基础知识: 1.
2024-03-04 10:02:13145 
 电子发烧友App
电子发烧友App









 工商网监
工商网监
评论