 将加速基因组分析扩展到RNA 、基因面板和注释
将加速基因组分析扩展到RNA 、基因面板和注释
NVIDIA Clara Parabricks v3 的发布。 6 去年夏天,在 全基因组和全外显子组测序分析综合工具包 中添加了多个加速体细胞变异调用者和用于注释和质量控制 VCF 文件的新工具。
在 2022 年 1 月发布的 Clara Parabricks v3 中。 7.NVIDIA 将工具包的范围扩展到新的数据类型,同时继续改进现有工具:
增加了对 RNASeq 分析的支持
通过加速实施 Fulcrum Genomics 的fgbio管道,增加了对基于 UMI 的基因面板分析的支持
增加了对mutect2正常面板( PON )过滤的支持,使加速mutectcaller符合 GATK 调用肿瘤正常样本的最佳实践
合并了一个bam2fq方法,该方法可以加速读取到新引用的重新对齐
使用ExpansionHunter增加了对短串联重复分析的支持
将呼叫后 VCF 分析步骤加快 15 倍
更新了HaplotypeCaller以匹配 GATK v4.1 ,并将DeepVariant更新为 v1.1
Clara Parabricks v3.7 显著拓宽了 Clara Parabricks 的功能范围,同时继续投资于领先的全基因组和全外显子组管道领域。
使参考基因组与 bam2fq 和 fq2bam 重新对齐
为了解决人类参考基因组的最新更新问题,并使重新排列读数便于大型研究, NVIDIA 开发了一种新的bam2fq工具。 Parabricks bam2fq可以从 BAM 文件中提取 FASTQ 格式的读取数据,为 GATK SamToFastq或bazam等工具提供了一个加速的替代品。
与 Parabricks fq2bam相结合,您可以使用八个 NVIDIA V100 GPU 在 90 分钟内将一个 30 倍 BAM 文件从一个引用(例如 hg19 )完全重新对齐到一个更新的引用( hg38 或 CHM13 )。内部基准测试表明,与仅依赖 hg19 相比,重新调整到 hg38 并重新运行变体调用可以在一瓶 HG002 真值集中捕获基因组中数千个真正的阳性变体。
重新调整后的变体调用的改进几乎与最初与 hg38 一致。虽然这个工作流程以前是可行的,但它的速度非常慢。 NVIDIA 最终将参考基因组更新应用于 Clara Parabricks 中最大的 WGS 研究。
| ############# | |
| ## Download the 30X hg19-aligned bam from Google's public sequencing of HG002 | |
| ## and the respective BAI file. | |
| ############# | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam.bai | |
| ############# | |
| ## Prepare the references so we can realign reads | |
| ############# | |
| ## Download the original hg19 / hsd37d5 reference | |
| ## and create and FAI index | |
| wget ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz | |
| gunzip hs37d5.fa.gz | |
| samtools faidx hs37d5.fa | |
| ## Download GRCh38 | |
| wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| gunzip GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| ## Make a .fai index using samtools faidx | |
| samtools faidx GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Create the BWA indices | |
| bwa index GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Download the Gold Standard indels from 1kg to use as your known-sites file. | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz | |
| ## Also grab the tabix index for the file | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi | |
| ############ | |
| ## Run the bam2fq tool to extract reads from the BAM file | |
| ## Adjust the --num-threads argument to reflect the number of cores on your system. | |
| ## With 8 GPUs and 64 vCPUs this should take ~45 minutes. | |
| ############ | |
| time pbrun bam2fq \ | |
| --ref hs37d5.fa \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq \ | |
| --num-threads 64 | |
| ############## | |
| ## Run the fq2bam tool to align reads to GRCh38 | |
| ############## | |
| time pbrun fq2bam \ | |
| --in-fq HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --knownSites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \ | |
| --out-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt |
 by
by RNASeq 转录本定量和融合调用 Clara Parabricks 的更多选项
在 3.7 版中, Clara Parabricks 还添加了两个用于 RNASeq 分析的新工具。
转录本定量是对 RNASeq 数据进行的最有效的分析之一。 Kallisto 是一种基于伪比对的快速表达量化方法。虽然 Clara Parabricks 已经将 STAR 纳入了 RNASeq 比对,但 Kallisto 添加了一种补充方法,可以运行得更快。
融合调用是另一种常见的 RNASeq 分析。在 Clara Parabricks 3.7 中, Arriba 提供了第二种方法,用于根据星形对齐器的输出调用基因融合。与恒星聚变相比,阿里巴可以调用更多类型的事件,包括:
病毒整合位点
内部串联复制
全外显子重复
环状 RNA
涉及免疫球蛋白和 T 细胞受体位点的增强子劫持事件
内含子和基因间区域中的断点
Kallisto 和 Arriba 的加入使 Clara Parabricks 成为许多转录组分析的综合工具包。
简化和加速基因面板和 UMI 分析
虽然全基因组和全外显子组测序在研究和临床实践中越来越普遍,但基因面板在临床领域占据主导地位。
基因小组工作流程通常使用独特的分子标识符( UMI )连接到读取,以提高低频突变的检测极限。NVIDIA 加速了 Fulcrum Genomics fgbio UMI 管道,并将八步管道整合到 v3 中的单个命令中。 7 ,支持多种 UMI 格式。
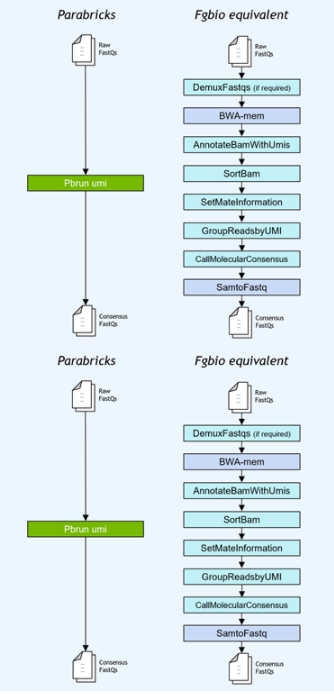
图 1 。 Fulcrum Genomics Fgbio-UMI 管道通过对 Clara Parabricks 的一个命令加速
使用 ExpansionHunter 检测短串联重复序列中的变化
短串联重复序列( STR )是某些神经系统疾病的公认原因,也是法医学和群体遗传学研究中指纹样本的重要标记。
NVIDIA 通过在 3.7 版中添加对ExpansionHunter的支持,在 Clara Parabricks 中实现了这些位点的基因分型。现在完全使用 Clara Parabricks 命令行界面就可以轻松地从原始读取转换为基因型 STR 。
利用 PON 支持改善静音体细胞突变通话
根据已知正常样本中的一组突变筛选体细胞突变调用是一种常见做法,也称为正常组( PON )。 NVIDIA 在mutectcaller工具中增加了对公共 PON 集和自定义 PON 的支持,该工具现在为体细胞突变呼叫提供了 GATK 最佳实践的加速版本。
加速呼叫后 VCF 注释和质量控制
在 v3 中。在第 6 版中, NVIDIA 添加了vbvm、vcfanno、frequencyfiltration、vcfqc和vcfqcbybam工具,使呼叫后 VCF 合并、注释、过滤、过滤和质量控制更易于使用。
v3 。 7 版本通过完全重写vbvm、vcfqc和vcfqcbybam的后端对这些工具进行了改进,所有这些工具现在都更加健壮,速度提高了 15 倍。
| #!/bin/bash | |
| ######################## | |
| ## In this gist, we'll reuse the commands from our 3.6 tutorial to align reads and generate BAM files. | |
| ## Check out the full post at https://medium.com/@johnnyisraeli/accelerating-germline-and-somatic-genomic-analysis-of-whole-genomes-and-exomes-with-nvidia-clara-e3deeae2acc9 | |
| and Gists at: | |
| ## https://gist.github.com/edawson/e84b2785db75d3c0aea9cc6a59969d45#file-full_pipeline_and_data_prep_parabricks3-6-sh | |
| ## and | |
| ## https://gist.github.com/edawson/e84b2785db75d3c0aea9cc6a59969d45#file-step_1_align_reads_parabricks3-6-sh | |
| ########### | |
| ########### | |
| ## We'll run this tutorial on a GCP VM with 64 vCPUs, 240GB of RAM, and 8x NVIDIA V100 GPUs | |
| ## To save costs, you can also run this on a GCP VM with 32 vCPUS, 120GB of RAM, and 4x V100 GPUs | |
| ########### | |
| ## After aligning our reads, we'll rerun the variant calling stages of our past gist | |
| ## since we've updated the haplotypecaller and DeepVariant tools. We'll | |
| ## also run Strelka2 as an additional variant caller. | |
| ## | |
| ## After that, we'll merge our VCFs to generate a union callset and an intersection VCF | |
| ## with variants called by all three variant callers, annotate our new intersection VCF, | |
| ## and remove variants that fail certain criteria for population frequency. | |
| ## Finally, we'll run our vcfqc and vcfqcbybam tools to generate simple quality control reports. | |
| ############# | |
| ################ | |
| ## HaplotypeCaller | |
| ## This step should take roughly 15 minutes on our 8xV100 VM. | |
| ################ | |
| time pbrun haplotypecaller \ | |
| --ref ~/refs/Homo_sapiens_assembly38.fasta \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --in-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt \ | |
| --out-variants HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf | |
| ################ | |
| ## DeepVariant | |
| ## This step should take approximately 20 minutes on an 8xV100 VM | |
| ################ | |
| time pbrun deepvariant \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-variants HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf | |
| ############### | |
| ## Strelka | |
| ## This step should take ~10 minutes on a 64-core VM. | |
| ############### | |
| time pbrun strelka \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --in-bams HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.strelka \ | |
| --num-threads 64 | |
| ## Copy strelka results to current directory. | |
| cp HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.strelka.strelka_work/results/variants/variants.vcf.gz* . | |
| ## BGZIP and tabix-index the deepvariant VCFs | |
| bgzip -@16 HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf | |
| tabix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf.gz | |
| ## BGZIP and tabix index the haplotypecaller VCFs | |
| bgzip -@16 HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf | |
| tabix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf.gz | |
| ## Run the votebasedvcfmerger tool to generate a union and intersection VCF. | |
| time pbrun votebasedvcfmerger \ | |
| --in-vcf strelka:variants.vcf.gz \ | |
| --in-vcf deepvariant:HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.deepvariant.vcf.gz \ | |
| --in-vcf haplotypecaller:HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.pb.haplotypecaller.vcf.gz \ | |
| --min-votes 3 | |
| --out-dir HG002.realign.vbvm | |
| ## The HG002.realign.vbvm directory should now contain a | |
| ## unionVCF.vcf file with the union callset of HaplotypeCaller, Strelka, and DeepVariant | |
| ## and aa filteredVCF.vcf file with only calls produced by all three callers. | |
| ## Annotate the intersection VCF with gnomAD, ClinVar, 1000 Genomes | |
| ## Download our annotation VCFs and tabix indices | |
| wget https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz | |
| wget https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz.tbi | |
| wget https://storage.googleapis.com/gcp-public-data--gnomad/release/2.1.1/liftover_grch38/vcf/exomes/gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz | |
| wget https://storage.googleapis.com/gcp-public-data--gnomad/release/2.1.1/liftover_grch38/vcf/exomes/gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz.tbi | |
| wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz | |
| wget http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20181203_biallelic_SNV/ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz.tbi | |
| ## Download an Ensembl GTF to annotate the VCF file with gene names | |
| wget http://ftp.ensembl.org/pub/release-105/gtf/homo_sapiens/Homo_sapiens.GRCh38.105.gtf.gz | |
| ## Unzip the GTF file and add the "chr" prefix to the chromosome names (Ensembl excludes this prefix by default. | |
| gunzip Homo_sapiens.GRCh38.105.gtf.gz | |
| awk '{if($0 !~ /^#/) print "chr"$0; else print $0}' Homo_sapiens.GRCh38.105.gtf > Homo_sapiens.GRCh38.105.chr.gtf | |
| time pbrun snpswift \ | |
| --input-vcf HG002.realign.vbvm/filteredVCF.vcf \ | |
| --anno-vcf 1000Genomes:ALL.wgs.shapeit2_integrated_v1a.GRCh38.20181129.sites.vcf.gz \ | |
| --anno-vcf gnomad_v2.1.1:gnomad.exomes.r2.1.1.sites.liftover_grch38.vcf.bgz \ | |
| --anno-vcf ClinVar:clinvar.vcf.gz \ | |
| --ensembl Homo_sapiens.GRCh38.105.chr.gtf \ | |
| --output-vcf HG002.realign.3callers.annotated.vcf | |
| ################## | |
| ## frequencyfiltration | |
| ## Next we'll filter our VCF to remove variants with 1000Genomes allele frequency > 0.05 | |
| ## and gnomAD AF < 0.05 | |
| ################## | |
| time pbrun frequencyfiltration \ | |
| --in-vcf HG002.realign.3callers.annotated.vcf \ | |
| --and-expression "1000Genomes_AF < 0.05" \ | |
| --and-expression "gnomad_v2.1.1_AF < 0.05" \ | |
| --out-vcf HG002.realign.3callers.annotated.filtered.vcf | |
| ################## | |
| ## Finally, we'll run our automated vcfqc tool to generate some | |
| ## basic QC stats. The vcfqcbybam tool could also be run | |
| ## to produce QC stats using an auxilliary BAM file (e.g., when variant calls don't have the desired fields). | |
| ################## | |
| time pbrun vcfqc --in-vcf HG002.realign.3callers.annotated.filtered.vcf \ | |
| --output-dir HG002.realign.3callers.annotated.filtered.qc \ | |
| --depth haplotypecaller_DP --allele-depth deepvariant_AD |
总结
带有 Clara Parabricks v3 。 7 .NVIDIA 致力于使 Parabricks 成为加速基因组数据分析的最全面解决方案。它是 WGS 、 WES 和现在的 RNASeq 分析以及基因面板和 UMI 数据的广泛工具包。
关于作者
Eric Dawson 是一位生物信息学科学家,他开发了种系和体细胞基因组分析的加速方法。在加入 NVIDIA 之前, Dawson 博士在剑桥大学和国家癌症研究所完成了博士学位。
Johnny Israeli 是NVIDIA 基因组学和药物发现软件的经理。他在斯坦福大学获得了博士学位,由 Anshul Kundaje 担任顾问,他的论文专注于基因组学的深入学习。他拥有勘萨斯大学物理学硕士和数学学士学位。
审核编辑:郭婷
| ############# | |
| ## Download the 30X hg19-aligned bam from Google's public sequencing of HG002 | |
| ## and the respective BAI file. | |
| ############# | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam | |
| wget https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch37/bam/hiseqx/wgs_pcr_free/30x/HG002.hiseqx.pcr-free.30x.dedup.grch37.bam.bai | |
| ############# | |
| ## Prepare the references so we can realign reads | |
| ############# | |
| ## Download the original hg19 / hsd37d5 reference | |
| ## and create and FAI index | |
| wget ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz | |
| gunzip hs37d5.fa.gz | |
| samtools faidx hs37d5.fa | |
| ## Download GRCh38 | |
| wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| gunzip GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz | |
| ## Make a .fai index using samtools faidx | |
| samtools faidx GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Create the BWA indices | |
| bwa index GCA_000001405.15_GRCh38_no_alt_analysis_set.fna | |
| ## Download the Gold Standard indels from 1kg to use as your known-sites file. | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz | |
| ## Also grab the tabix index for the file | |
| wget https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi | |
| ############ | |
| ## Run the bam2fq tool to extract reads from the BAM file | |
| ## Adjust the --num-threads argument to reflect the number of cores on your system. | |
| ## With 8 GPUs and 64 vCPUs this should take ~45 minutes. | |
| ############ | |
| time pbrun bam2fq \ | |
| --ref hs37d5.fa \ | |
| --in-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam \ | |
| --out-prefix HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq \ | |
| --num-threads 64 | |
| ############## | |
| ## Run the fq2bam tool to align reads to GRCh38 | |
| ############## | |
| time pbrun fq2bam \ | |
| --in-fq HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq_1.fastq.gz \ | |
| --ref Homo_sapiens_assembly38.fasta \ | |
| --knownSites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \ | |
| --out-bam HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.bam \ | |
| --out-recal-file HG002.hiseqx.pcr-free.30x.dedup.grch37.bam2fq.hg38.BQSR-REPORT.txt |
RN
-
NVIDIA
+关注
关注
14文章
4983浏览量
103002 -
gpu
+关注
关注
28文章
4730浏览量
128904
发布评论请先 登录
相关推荐
AI for Science:人工智能驱动科学创新》第4章-AI与生命科学读后感
【飞凌嵌入式OK3576-C开发板体验】RKNPU图像识别测试
通过应用频率将TPS92210的调光范围扩展到通用AC范围

NVIDIA Parabricks v4.3.1版本的新功能
微远基因宣布完成数亿元人民币D+轮融资,领跑病原分子精准诊断赛道
威世将凯睿德制造 MES 扩展到半导体业务
罗德与施瓦茨推出频率可达50GHz专用相位噪声分析的FSPN50
一种用于微液滴中单细胞无标记分析的液滴筛选(LSDS)方法
基于上转换纳米颗粒的侧流免疫平台,用于超灵敏检测miRNA
高通量测序技术及原理介绍
芯动力再创辉煌:M.2 AI加速卡赢得海外知名企业青睐!






 工商网监
工商网监
评论