 卤化物电解质阴离子六方密堆框架下普适的相结构调控与锂离子传导行为
卤化物电解质阴离子六方密堆框架下普适的相结构调控与锂离子传导行为

基于无机固态电解质的全固态锂电池由于其高安全性和高能量密度被认为是较为先进的储能系统。全固态锂离子电池由于其较高的理论能量密度及高离子电导率固态电解质的快速发展而被认为极具潜力的储能体系之一。卤化物电解质具有较宽的电化学窗口、优良的空气稳定、与电极材料较好兼容性等优点。其中,Li3M(III)Cl6构型的卤化物电解质有三种结构,包括单斜相(C2/m),三方相(P-3m1)和正交相(Pnma)。通常认为中M3+半径较小(如Yb,Lu等)的Li3M(III)Cl6形成正交相,而M3+半径较大 (如Tb, Dy, Ho, Er, Tm等)的Li3M(III)Cl6则形成三方相。研究不同晶体结构、组份及其对锂离子电导率的影响对合理设计发展高离子电导率、高稳定性的卤化物固态电解质是非常重要的。
【工作介绍】
近日,加拿大西安大略大学孙学良教授课题组,与荷兰代尔夫特理工大学Marnix Wagemaker教授课题组合作,基于先前卤化物电解质的工作,进一步发现、合成并揭示了在Li-M-Cl(M = Dy、Ho、Y、Er、Tm)体系中有利的正交晶相。从三方到正交晶相的转变能够显著提高其锂离子电导率并降低锂离子扩散的活化能垒。该研究结果有望发现稀土金属卤化物的基本化学理论,探索具有高锂离子电导率的新材料。相关研究成果以“A series of ternary metal chloride superionic conductors for high-performance all-solid-state lithium batteries”为题发表在国际顶级期刊Adv. Energy. Mater.上。梁剑文为本文第一作者。
【内容表述】
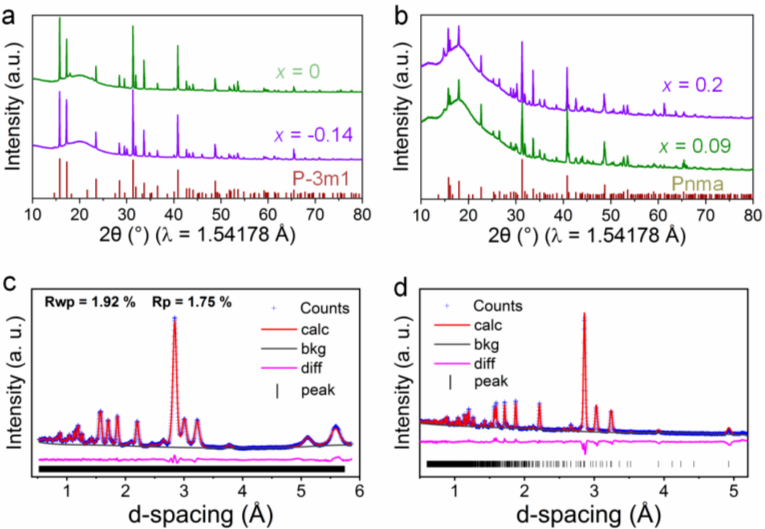
Figure 1. Li-Ho-Cl体系的XRD和中子衍射图。(a) Li3HoCl6(x = 0)和Li3.42Ho0.86Cl6(x = -0.14)样品的XRD图谱,均为P-3m1空间群。(b) Li2.4Ho1.2Cl6(x = 0.2) 和 Li2.73Ho1.09Cl6(x = 0.09) 样品的 XRD 图谱,均为Pnma 空间群。(c) Li3HoCl6和(d) Li2.73Ho1.09Cl6样品的中子衍射图普及Rietveld拟合。
以Li-Ho-Cl体系为例,通过共融法以LiCl和HoCl3为原料在650℃下制备了一系列的Li3-3xHo1+xCl6电解质材料。当x=0和x=-0.14时,得到的样品分别为Li3HoCl6和Li3.42Ho0.86Cl6,XRD衍射分析表明这两个样品均为三方P-3m1结构;当x=0.2和x=0.09时,得到的样品分别为 Li2.4Ho1.2Cl6和 Li2.73Ho1.09Cl6,XRD衍射分析表明这两个样品均为正交Pnma结构。对Li3HoCl6和Li2.73Ho1.09Cl6样品进一步的中子衍射分析同样证明了此两种材料的不同结构。
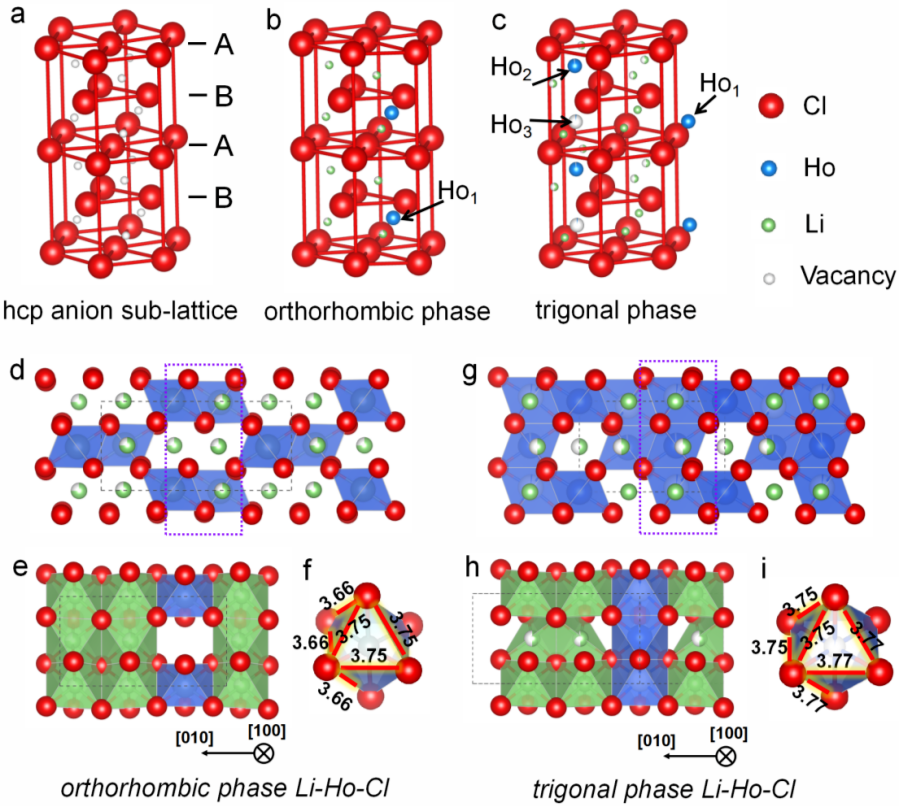
Figure 2. Li3HoCl6的正交和三方结构的阴离子/阳离子排列。(a-c) 正交和三方结构中Ho和空位的不同阳离子排列。(d, e) Li3HoCl6晶胞的正交结构。(f) Li3HoCl6正交晶相中的 HoCl63-八面体。(g, h) Li3HoCl6晶胞的三方结构。(i) Li3HoCl6三方相中的 HoCl63-八面体。绿色,Li;蓝色,Ho;红色,Cl。
正交和三方相的Li-Ho-Cl电解质均是基于Cl-阴离子的hcp堆积结构,六个Cl-组成的八面体被Ho3+, Li+,或者空位占据。正交和三方相的主要区别在于阳离子在这些Cl-八面体的占位,如图2d-h所示。在正交 Pnma 结构中,4c 位点被 Ho3+完全占据。Li1 和 Li2 分布在 8d 位点,占用参数分别为 0.848 和 0.652。每个 HoCl63-八面体被三个共享边缘的 LiCl65-八面体包围(图 2d,e)。在三方结构中,存在三个 Ho 位点(图 2c)。一个被完全占据(Ho1,1a 位点),另外两个被部分占据。沿 c 轴有两个 Li 位点,完全占据的 Li1 层(6g 位点)和半占据的 Li2 层(6d 位点)。每个 HoCl63-八面体被六个共享边缘的 LiCl65-八面体包围,在 ab 平面上形成蜂窝晶格(图 2g,h)。三角结构在[001]方向上包含无限的共享面HoCl63-八面体链。HoCl63-八面体的局部结构在两种结构之间也不同(图2f,i),不对称的局部环境会在单斜相的 Li3HoCl6中具有由非对称性引起的扭曲的Cl-排列(图 2f) 。
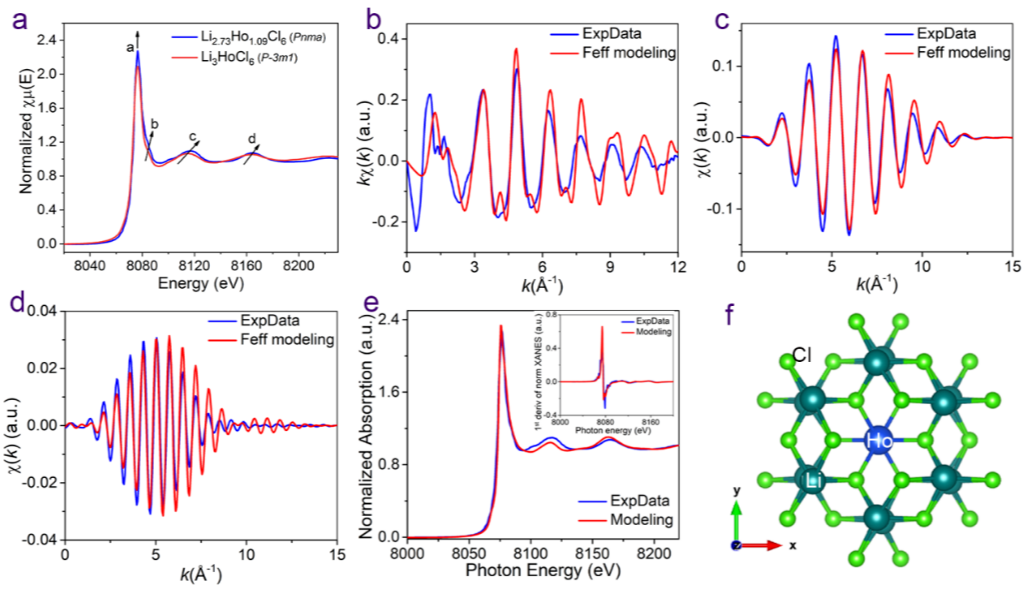
Figure 3. XAFS拟合的Li-Ho-Cl中 Ho 的局部环境。(a) Pnma 相Li2.73Ho1.09Cl6和 P-3m1 相Li3HoCl6样品的Ho L3-edge的XAFS光谱。(b) Ho在Li2.73Ho1.09Cl6(Pnma)的局部环境的k空间谱图(实验数据为蓝线,Feff建模为红线)。(c) Li2.73Ho1.09Cl6(Pnma) 的第一壳层经傅里叶转换的k空间谱图。(d) Li2.73Ho1.09Cl6(Pnma) 的第二壳层傅里叶转换的k空间谱图。(e) XANES谱实验(蓝线)与理论模型的最佳拟合(红线)之间的比较。(f) 第一和第二壳层Ho周围的结构(以Ho为中心,半径为 R = 6Å)。
对合成的Pnma相Li2.73Ho1.09Cl6和P-3m1相Li3HoCl6样品进行X 射线吸收精细和X 射线吸收近边缘结构分析。Pnma和 P-3m1相之间Ho局部环境的差异来自于 Pnma 相中HoCl63-八面体的轻微变形以及与Li和空位有关的第二层中阳离子的不同排列。Li2.73Ho1.09Cl6(Pnma) 的第一壳FT的整体kχ(k)、反傅立叶变换 (BFT) 滤波kχ(k)和第二壳FT的BFT滤波kχ(k)实验数据和Feff模型的基本一致,进一步的R 空间曲线拟合与初始 Feff 建模也一致。因此,通过初始 Feff 建模、R空间曲线拟合和 XANES 理论建模证明了Li2.73Ho1.09Cl6八面体位点的Ho占据,进一步验证了XRD和中子细化得出的结构准确性。
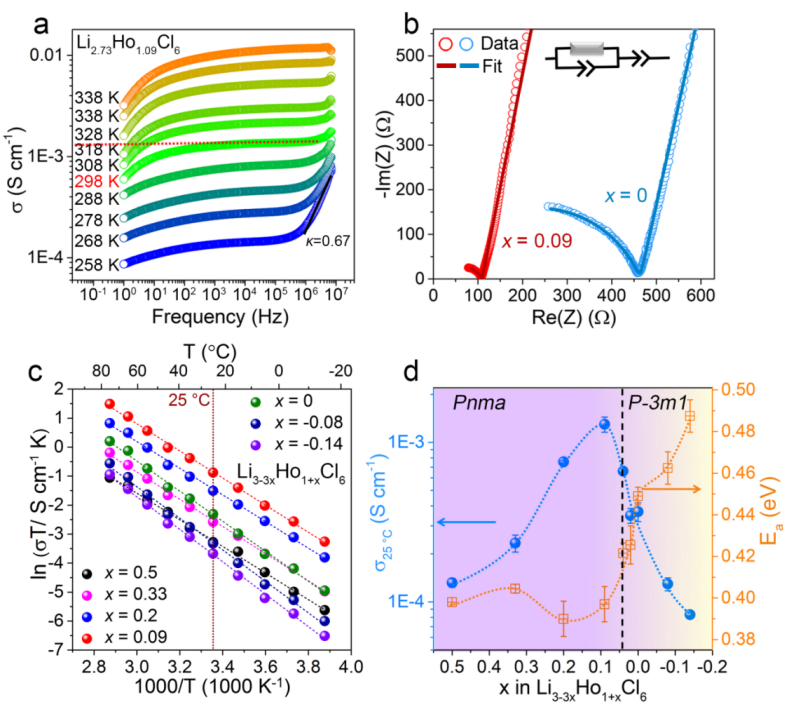
Figure 4. (a) 在不同温度下的 Li2.73Ho1.09Cl6电解质的电导率等温线 σ(ν)。(b) 制备的Pnma相Li2.73Ho1.09Cl6和P-3m1相Li3HoCl6电解质在25 ℃时的EIS谱。(c) Li3-3xHo1+xCl6电解质的Arrhenius图 (-0.14 ≤ x ≤ 0.5)。(d) Li3-3xHo1+xCl6电解质的室温离子电导率和相应的活化能 (-0.14 ≤ x ≤ 0.5)。
交流阻抗测试表明,对于Pnma结构 (0.04 < x ≤ 0.2)的Li3-3xHo1+xCl6电解质,其室温离子电导率随着x的减小而逐渐增加,其中在x = 0.09 时,即Li2.73Ho1.09Cl6具有最高的离子电导率,为1.3 × 10-3 S cm-1。当进一步降低x ≤ 0.02 时,Li3-3xHo1+xCl6从 Pnma 变为 P-3m1,离子电导率显着降低。当Li3HoCl6 (P-3m1) 的室温离子电导率为2.9 × 10-4 S cm-1。此外,Li3-3xHo1+xCl6电解质的活化能呈现相反的趋势(图 4d)。具有Pnma相的Li3-3xHo1+xCl6电解质的活化能低于0.4 eV,对于具有P-3m1相的Li3-3xHo1+xCl6电解质,该值随着x的减小而显着增加。这表明与P-3m1相比,Pnma对称性的阳离子排列支持较低的Li+传输活化能。
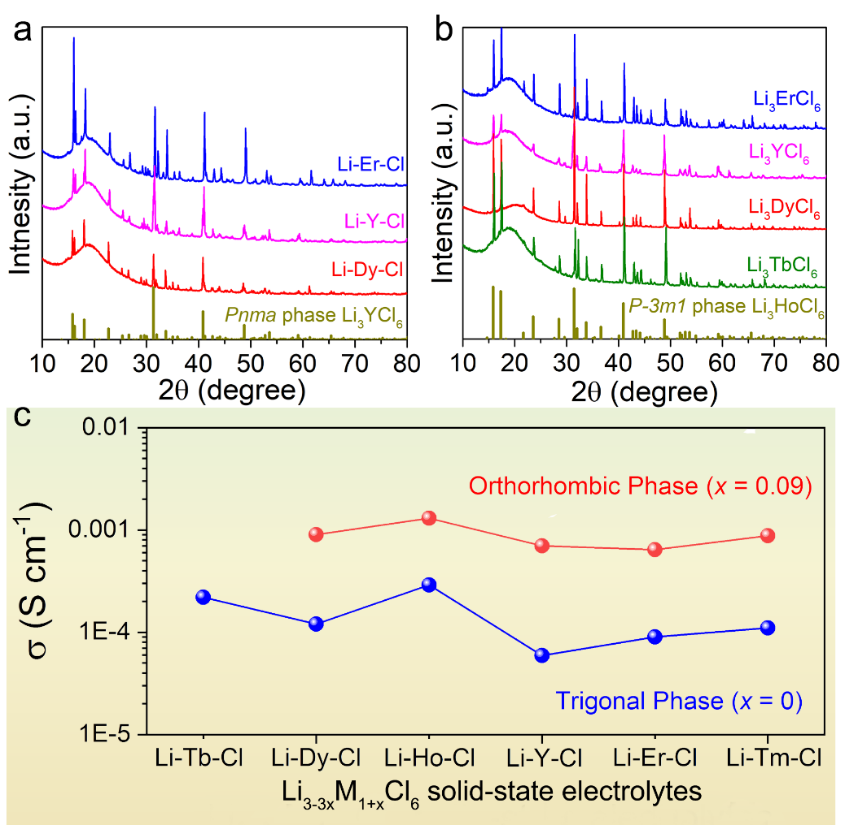
Figure 5. (a) Li2.73M1.09Cl6(x = 0.09, M = Er, Y, Dy, Pnma 相) 的XRD图谱。(b) Li3MCl6(x = 0, M = Er, Y, Dy, Tb, P-3m1相) 的XRD图谱。(c) Li3-3xHo1+xCl6SSE 的室温离子电导率。
此外,已经发现,除了Li-Tb-Cl组分之外,在其他类型的 Li-M(III)-Cl SSE 中,三方相到正交相随着组份变化的相变是普遍的。虽然之前已经报道了Li3YCl6的亚稳态正交结构,但对于其他Li-M(III)-Cl (M = Ho, Er, Dy, Tm) 材料来说,本工作中第一次实现正交相,这使得对三元稀土氯化物的化学结构有了更清晰的认识。同时,在所有 Li3−3xM1+xCl6电解质中都可以发现从三方相到正交相引起的离子电导率增加。正交相的Li2.73Dy1.09Cl6、Li2.73Y1.09Cl6、Li2.73Er1.09Cl6、Li2.73Tm1.09Cl6的室温离子电导率分别为 9.0×10-4、7.0×10-4、6.4×10-4和8.9 × 10-4S cm-1,比具其对应的三方相的 Li3MCl6离子电导率高一个数量级。因此,Li3−3xM1+xCl6此类材料中的结构转变过及离子电导率变化展示出一种普遍现象,可用于探索和实现许多新材料。
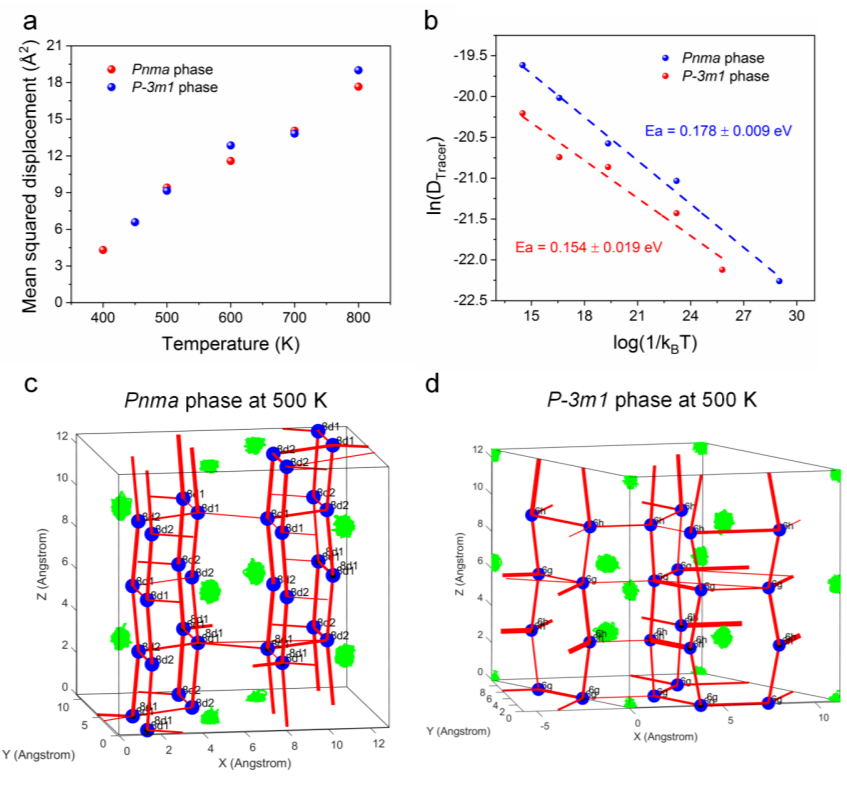
Figure 6. (a) 400-800K 的均方位移 (MSD)。(b) 不同温度下扩散系数与1/kbT的对数。(c) Pnma相和 (d) P-3m1相在500K时不同Li位点之间的离子跳跃。锂位点(蓝色)、Ho轨迹(绿色)和锂离子跳跃(红色)。Li位点的大小和线的粗细分别代表模拟中该位点的占用率和跳跃频率。
进一步通过从头算分子动力学对Li3HoCl6的组成对两相进行了模拟。两种结构从Arrhenius图的斜率得到的活化能几乎相似,与实验结果略有不同。不同温度下不同锂位点之间的跳跃分析表明,对于500 K的正交相结构,在 Wyckoff 8d1 和 8d2 位点之间沿 z 方向存在清晰的长程扩散路径。在xy平面中 8d1-8d1 和8d2-8d2位点之间的跳跃也是可能的,但这些跳跃的频率要低得多;而沿 z 方向的跳跃比跨平面跳跃具有更低的活化能,表明高电导率主要是由于这些一维扩散路径。对于三方相结构,沿z方向跳跃的跳跃位于6h和6g位点之间,并且发生的频率(39.3~51.6%)与相同Wyckoff位点之间的跳跃(48.4~60.7%)大致相同;连接六边形的跳跃几率较小(没有线条或非常细的红线),这表明Li+的长程扩散和可能沿 z 方向的路径最有利,但与正交相相相比,发生跳跃的频率相对较低。结果表明,正交相中z方向的扩散更有利于实现高电导率。
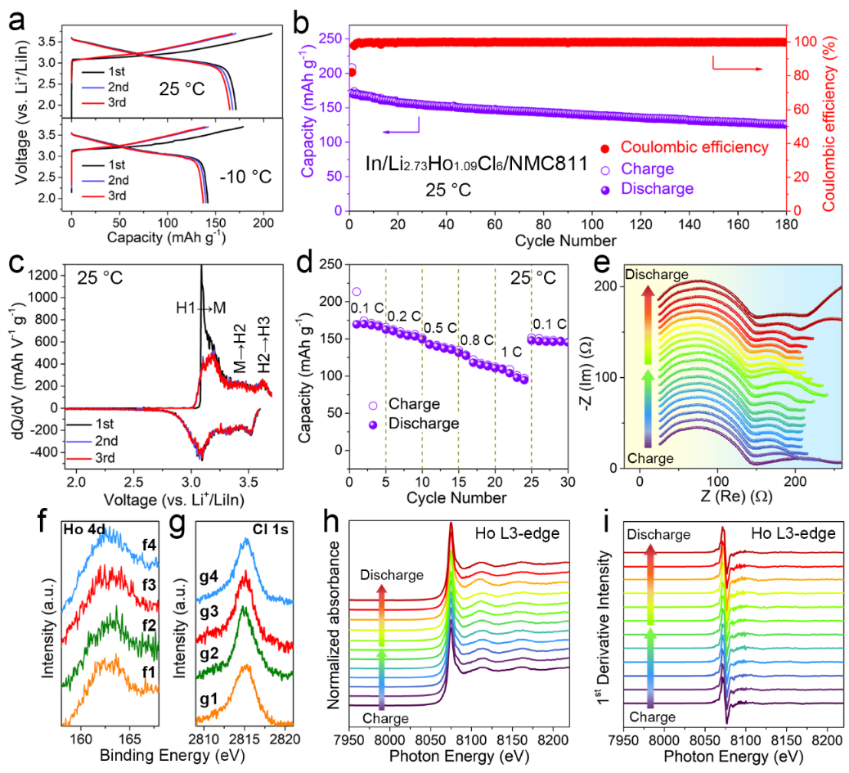
Figure 7. 全固态NMC811/Li2.73Ho1.09Cl6/In 电池的电化学性能。(a) 充电/放电曲线和 (b) 循环性能 (25℃, 0.1 C)。(c) 前三圈的 dQ/dV 曲线。(d) 倍率性能。(e) 全固态NMC811/Li2.73Ho1.09Cl6/In电池在充电/放电2小时和静置2小时后进行的 EIS 光谱。(f, g) NMC811-Li2.73Ho1.09Cl6正极在光子能量3000 eV下的 Ho4dCl1s 光谱在不同充电/放电状态下的HEXPS。NMC811-Li2.73Ho1.09Cl6正极在不同充放电状态下的(h)Ho L3-edge和(i)Ho L3-edge XANES光谱的一阶导数。
将Li2.73Ho1.09Cl6卤化物固态电解质直接与商业NMC811混料得到正极材料。电化学性能测试表明,NMC811/Li2.73Ho1.09Cl6/In固态电池具有较高的首圈库伦效率,较好的循环稳定性及倍率性能。充放电过程中的阻抗测试、高能 X 射线光电子能谱及Ho L3-edge的XANES光谱表明,Li2.73Ho1.09Cl6-NMC811具有较稳定的界面,Li2.73Ho1.09Cl6电解质在整个充放电过程是稳定的。
【结论】
该工作揭示了三元金属氯化物固体电解质Li3-3xM1+xCl6(-0.14 < x ≤ 0.2, M = Tb, Dy, Ho, Y, Er, Tm)随着x值增加导致的三方相到正交相的转变。通过EIS、X射线和中子衍射以及MD模拟理论计算揭示了锂离子电导率与结构之间的关系。对于正交相Li-Ho-Cl、Li-Dy-Cl、Li-Y-Cl、Li-Er-Cl 和 Li-Tm-Cl,均展示较高的锂离子电导率(比三方相的结构高一个数量级)。MD模拟理论计算表明正交相结构中z方向的锂离子传输对其离子电导率的提升有显著的作用。NMC811/Li2.73Ho1.09Cl6/In全固态电池在室温和低温下均表现出优异的电化学性能。这些结果为新型卤化物超离子导体的设计提供了指导。
审核编辑 :李倩
-
锂离子
+关注
关注
5文章
572浏览量
39962 -
电解质
+关注
关注
6文章
841浏览量
21560 -
电导率
+关注
关注
1文章
308浏览量
15646
原文标题:西安大略大学孙学良团队AEM: 卤化物电解质阴离子六方密堆框架下普适的相结构调控与锂离子传导行为
文章出处:【微信号:Recycle-Li-Battery,微信公众号:锂电联盟会长】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
突破固态电解质瓶颈!我国研发新型金属有机钾离子导体

Avio 200 ICP-OES测定固态电解质中杂质元素含量

高粘度聚合物电解质调控锂沉积模式:助力高性能固态锂金属电池

氮化硅陶瓷赋能LLZO固态电解质:界面相容性研究与产业化前景

高成本难题破解:新型非晶态卤化物固态电解质引领行业变革
纳米结构对齐复合固态电解质:全固态电池离子传输与界面接触新突破
国内企业突破固态锂电池电解质瓶颈
全固态锂电革命:垂直取向超离子通道复合电解质的创新突破
巴西研究团队推进钠离子电池电解质计算研究

固态电池新突破!新能源车续航有望翻倍
突破性固态聚合物电解质:像拼图一样组装分子,打造安全高压锂电池
共聚焦显微镜观测:电解质等离子抛光工艺后的TC4 钛合金三维轮廓表征

锂离子电池电解质填充工艺:技术原理与创新实践
锂离子电池创:性能、分类与GPE的应用前景
锂离子电池电解液浸润机制解析:从孔隙截留到工艺优化



评论