 一种量子化学与分子力学结合的方法
一种量子化学与分子力学结合的方法
本专题将介绍一种量子化学与分子力学结合的方法(QM/MM方法),该方法既包括量子化学的精确性,又利用分子力学的高效性,其基本思想是用量子力学处理感兴趣的区域,其余部分用经典分子力学来处理。本章节以一个典型的没食子酸分子(Gallic Acid,GA)为例,介绍输入文件准备,QM/MM单点计算,QM/MM结构优化,QM/MM激发态计算。其中,BDF程序主要完成量子化学计算部分,其余部分由BDF开发成员修改的pDynamo2.0程序包完成。同时介绍如何读取数据用于结果分析,帮助用户深入了解BDF软件的使用。
输入文件准备
一般来说,QM/MM计算之前,需要对目标体系进行分子动力学模拟,得到适合的初始构象。当采用PDB、MOL2或xyz文件作为输入时,pDynamo2.0程序包仅支持OPLS力场,对于小分子和非标准氨基酸力场参数不全,不推荐使用。建议优先采用Amber程序,通过拓扑文件输入力场参数。以Amber为例,从动力学模拟轨迹提取感兴趣的结构存储于.crd文件中,与对应的参数/拓扑文件.prmtop一起可以作为QM/MM计算的起始点。Python脚本如下:
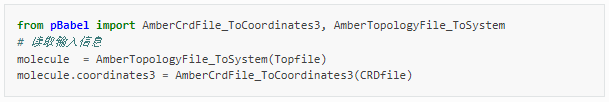
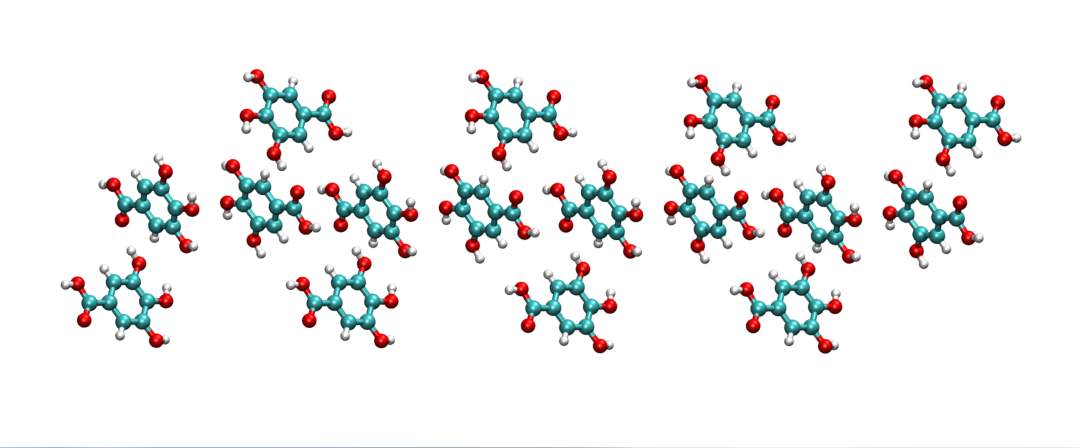
其中,需要提前安装好AmberTools,python2.0版本,并正确设置好AMBERHOME和PDYNAMO环境变量,关于如何将GallicAcid.pdb初始结构文件(图1,晶胞为2*1*1)生成使用AmberTools21程序相对应的坐标文件GallicAcid.crd和参数/拓扑文件GallicAcid.prmop的方法如下:
运行antechamber程序将Pdb文件转化为mol2文件:antechamber -i GallicAcid.pdb -fi pdb -o GallicAcid.mol2 -fo mol2 -j 5 -at amber -dr no
-i 指定输入文件
-fi 指定输入文件类型
-o 指定输出文件
-fo 指定输出文件类型
-j匹配原子类型和键类型
-at定义原子类型
运行parmchk2程序生成对应体系的力场参数文件:parmchk2 -i GallicAcid.mol2 -f mol2 -o GallicAcid.frcmod
运行tleap程序构建系统拓扑并为分子定义力场参数步骤如下:
1.使用tleap命令启动tleap程序

2. 确定并加载体系力场:source leaprc.gaff(此为GAFF力场)
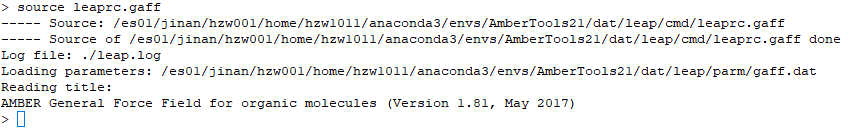
3. 调入配体mol2文件:GA = loadmol2 GallicAcid.mol2

4. 检查导入的结构是否准确或缺失参数:check GA
5. 调入体系分子的模板,并补全库文件中缺失的参数:loadamberparams GallicAcid.frcmod
6. 准备生成的Sustiva库文件:saveoff GA GallicAcid.lib
7. 修改生成的Sustiva库文件并调入该文件:loadoff GallicAcid.lib

8. 保存.crd和.prmop文件:saveamberparm GA GallicAcid.prmtop GallicAcid.crd

9. 退出tleap程序:quit
分子动力学模拟
1.此处采用amber软件进行分子动力学模拟,首先对体系进行能量最小化模拟,输入文件min.in如下:
Initial minimisation of GallicAcid complex
&cntrl
imin=1, maxcyc=200, ncyc=50,
cut=16, ntb=0, igb=1,
&end
imin=1:运行能量最小化
maxcyc=200:能量最小化的最大循环数
ncyc=50:最初的0到ncyc循环使用最速下降算法, 此后的ncyc到maxcyc循环切换到共轭梯度算法
cut=16:以埃为单位的非键截断距离
ntb=0:关闭周期性边界条件
igb=1:Born模型
使用如下命令运行能量最小化:
sander -O -i min.in -o GallicAcid_min.out -p GallicAcid.prmtop -c GallicAcid.crd -r GallicAcid_min.rst &
其中GallicAcid_min.rst为输出包含坐标和速度的重启文件
2.接下来利用最小化模拟得到的重启文件升温系统,从而完成分子动力学模拟,输入文件md.in如下:
Initial MD equilibration
&cntrl
imin=0, irest=0,
nstlim=1000,dt=0.001, ntc=1,
ntpr=20, ntwx=20,
cut=16, ntb=0, igb=1,
ntt=3, gamma_ln=1.0,
tempi=0.0, temp0=300.0,
&end
imin=0:进行分子动力学(MD)
irest=0:读取先前保存的重新启动文件读取坐标和速度
nstlim=1000:运行的MD步数
dt=0.001:时间步长(单位:ps)
ntc=1:不启用SHAKE约束
ntpr=20:每ntpr步输出能量信息mdout一次
ntwx=20:每ntwx步输出Amber轨迹文件mdcrd一次
ntt=3:Langevin恒温器控制温度
gamma_ln=1.0:Langevin恒温器的碰撞频率
tempi=0.0:模拟的初始温度
temp0=300.0:模拟的最终温度
使用如下命令运行分子动力学模拟:
sander -O -i md.in -o md.out -p GallicAcid.prmtop -c GallicAcid_min.rst -r GallicAcid_md.rst -x GallicAcid_md.mdcrd -inf GallicAcid_md.mdinfo
其中GallicAcid_md.mdcrd文件即为MD模拟的轨迹文件,可借助VMD软件进行可视化显示分子结构,并从动力学模拟轨迹提取感兴趣的结构存储于.crd文件中。
QM/MM总能量计算
分子动力学模拟后提取文件为GallicAcid.prmtop, GallicAcid.crd,可对体系进行全量子化学总能量计算,python代码如下:
import glob, math, os
from pBabel import AmberCrdFile_ToCoordinates3, AmberTopologyFile_ToSystem
from pCore import logFile
from pMolecule import QCModelBDF, System
# 读取水盒子坐标和拓扑信息
molecule = AmberTopologyFile_ToSystem ("GallicAcid.prmtop")
molecule.coordinates3 = AmberCrdFile_ToCoordinates3("GallicAcid.crd")
# 定义能量计算模式,此处为全体系密度泛函计算,可以定义方法和基组,分别为GB3LYP和6-31g,
model = QCModelBDF("GB3LYP:6-31g")
molecule.DefineQCModel(model)
molecule.Summary() #输出体系计算设置信息
# 计算总能量
energy = molecule.Energy()
除了可以用全量子化学QM计算体系总能量,也可对感兴趣的分子进行QM/MM计算(本例为指定第五个分子用QM方法计算),QM/MM组合能量计算python脚本如下:
import glob, math, os
from pBabel import AmberCrdFile_ToCoordinates3, AmberTopologyFile_ToSystem
from pCore import logFile, Selection
from pMolecule import NBModelORCA, QCModelBDF, System
# 定义能量计算模式
nbModel = NBModelORCA() #处理QM和MM区相互作用
qcModel = QCModelBDF("GB3LYP:6-31g")
# 读取体系坐标和拓扑信息
molecule = AmberTopologyFile_ToSystem("GallicAcid.prmtop")
molecule.coordinates3 = AmberCrdFile_ToCoordinates3("GallicAcid.crd")
# 关闭体系对称性
molecule.DefineSymmetry(crystalClass = None) #QM/MM方法不支持使用周期性边界条件,故关闭周期性边界条件
# 指定QM区
qm_area = Selection.FromIterable(range (72, 90)) # 指定第五个分子用QM方法计算,其中(72, 90)指明原子列表索引值为72,73,74…..87,88,89,该值=原子序数-1
# 定义能量计算模式
molecule.DefineQCModel (qcModel, qcSelection = qm_area)
molecule.DefineNBModel (nbModel)
molecule.Summary()
# 计算总能量
energy = molecule.Energy()
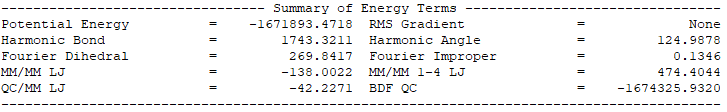
QM/MM模拟的输出总结了MM部分,QM部分,QM区和MM区相互作用部分的计算细节如下:

输出体系总能量信息以及各部分能量贡献如下:
-
软件
+关注
关注
69文章
4958浏览量
87626 -
程序
+关注
关注
117文章
3788浏览量
81112 -
动力学
+关注
关注
0文章
105浏览量
16979
原文标题:鸿之微BDF软件计算赏析|采用BDF的QM/MM多尺度计算方法研究晶体的光物理性质
文章出处:【微信号:hzwtech,微信公众号:鸿之微】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
【一文看懂】什么是量子计算?

一种高灵敏的化学检测系统
量子通信与量子计算的关系
菱形石墨烯结构及其中的量子反常霍尔效应
华为公开量子计算新专利
“本源悟空”超导量子计算机助力大规模流体动力学量子计算

基于量子干涉技术的单分子晶体管问世
量子计算机应用-量子计算流体力学软件






 工商网监
工商网监
评论