 量化软件BDF的计算
量化软件BDF的计算

根据Kasha规则,由于高激发态之间存在快速的无辐射跃迁,分子的荧光或磷光的初始状态是最低的单重态或三重态。薁(azulene)是典型违反 Kasha 规则的例子。azulene的荧光来自于S2态,可以认为是由于S2→S1能隙比较大,所以降低了S2→S1内转换速率。另外,由于S1和S0之间的能隙相对较小,所以其内转换速率很大,从而降低了S1→S0的荧光量子效率,所以S1→S0的荧光很难被发现。这里以具有反常荧光现象的azulene为例,使用BDF软件和MOMAP软件,计算azulene的S1→S0的辐射速率和内转换速率,从而解释azulene第一激发态极低的量子效率导致其荧光难以被观测到的实验结果。
MOMAP对azulene的S1→S0的辐射速率和内转换速率的计算需要BDF量化软件的结构优化频率结果文件、非绝热耦合结果文件和输入参数的计算。首先介绍量化软件BDF的计算。
准备azulene分子结构的xyz文件如下:
18
C -0.48100000 0.74480000 0.00000000
C -0.56240000 -0.71320000 0.00020000
C -1.75790000 1.17860000 -0.00030000
C -1.96510000 -1.08880000 0.00000000
C 0.66870000 1.60890000 0.00030000
C 0.45100000 -1.58850000 0.00030000
C -2.66930000 0.05180000 -0.00010000
C 1.95140000 1.22210000 0.00020000
C 1.86730000 -1.29960000 -0.00020000
C 2.49720000 -0.11610000 -0.00040000
H -2.09080000 2.20560000 -0.00040000
H -2.35750000 -2.09240000 0.00010000
H 0.46620000 2.67860000 0.00070000
H 0.22090000 -2.65340000 0.00040000
H -3.74370000 0.14050000 -0.00030000
H 2.70720000 2.00650000 0.00050000
H 2.49320000 -2.19150000 -0.00060000
H 3.58670000 -0.13930000 -0.00080000
打开Device studio,点击File-new project,命名为fluorescence.hpf,将azulene.xyz拖入Project中,双击azulene.hzw,得到如图所示界面。
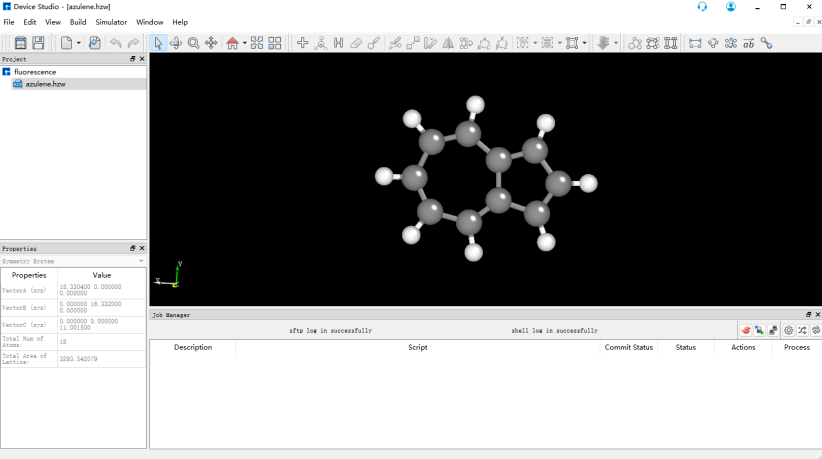
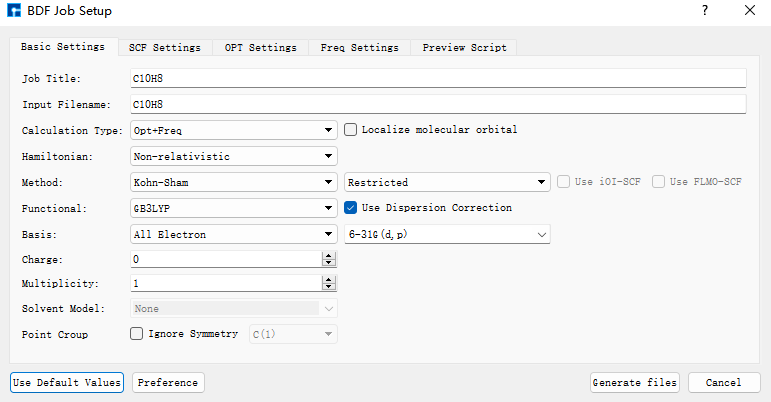
首先使用BDF进行azulene的结构优化和频率计算。选中Simulator → BDF → BDF,界面中设置参数。在Basic Settings界面中的Calculation Type选择Opt+Freq,方法采用默认的GB3LYP泛函,基组在Basis中的All Electron类型中,选择6-31G(d,p)。Basic Settings界面中的其它参数以及SCF Settings、OPT Settings、Freq Settings等面板的参数使用推荐的默认值,不需要做修改。之后点击 Generate files 即可生成对应计算的输入文件。
选中生成的bdf.inp文件,右击选择open with,打开该文件,如下所示:
$compass
Title
C10H8
Geometry
C -0.48100000 0.74480000 0.00000000
C -0.56240000 -0.71320000 0.00020000
C -1.75790000 1.17860000 -0.00030000
C -1.96510000 -1.08880000 0.00000000
C 0.66870000 1.60890000 0.00030000
C 0.45100000 -1.58850000 0.00030000
C -2.66930000 0.05180000 -0.00010000
C 1.95140000 1.22210000 0.00020000
C 1.86730000 -1.29960000 -0.00020000
C 2.49720000 -0.11610000 -0.00040000
H -2.09080000 2.20560000 -0.00040000
H -2.35750000 -2.09240000 0.00010000
H 0.46620000 2.67860000 0.00070000
H 0.22090000 -2.65340000 0.00040000
H -3.74370000 0.14050000 -0.00030000
H 2.70720000 2.00650000 0.00050000
H 2.49320000 -2.19150000 -0.00060000
H 3.58670000 -0.13930000 -0.00080000
End Geometry
Basis
6-31G(d,p)
Skeleton
Group
C(1)
$end
$bdfopt
Solver
1
MaxCycle
108
IOpt
3
Hess
final
$end
$xuanyuan
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
GB3LYP
D3
MPEC+COSX
Molden
$end
$resp
Geom
$end
选中bdf.inp文件,右击选择Run,弹出如下界面:
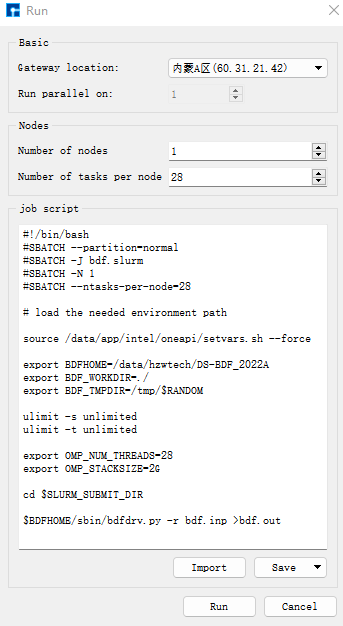
点击Run提交作业,会自动弹出结果文件的实时输出。
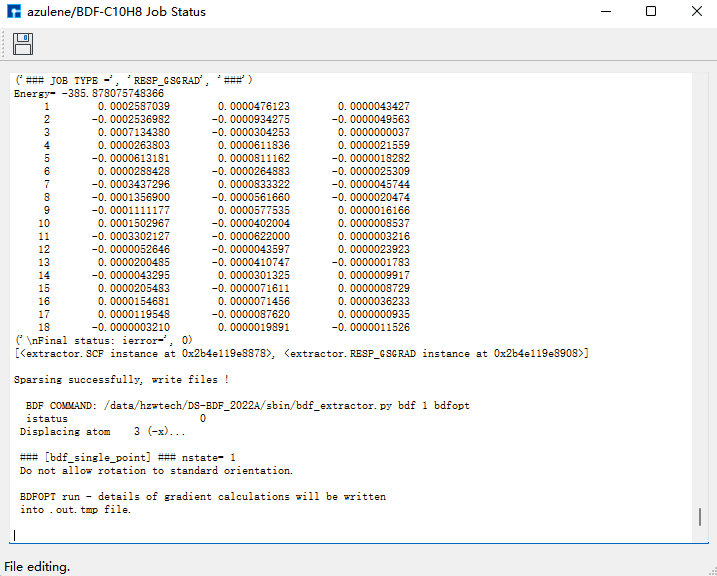
任务结束后bdf.out,bdf.out.tmp,bdf.scf.molden三个结果文件会出现在Project中。
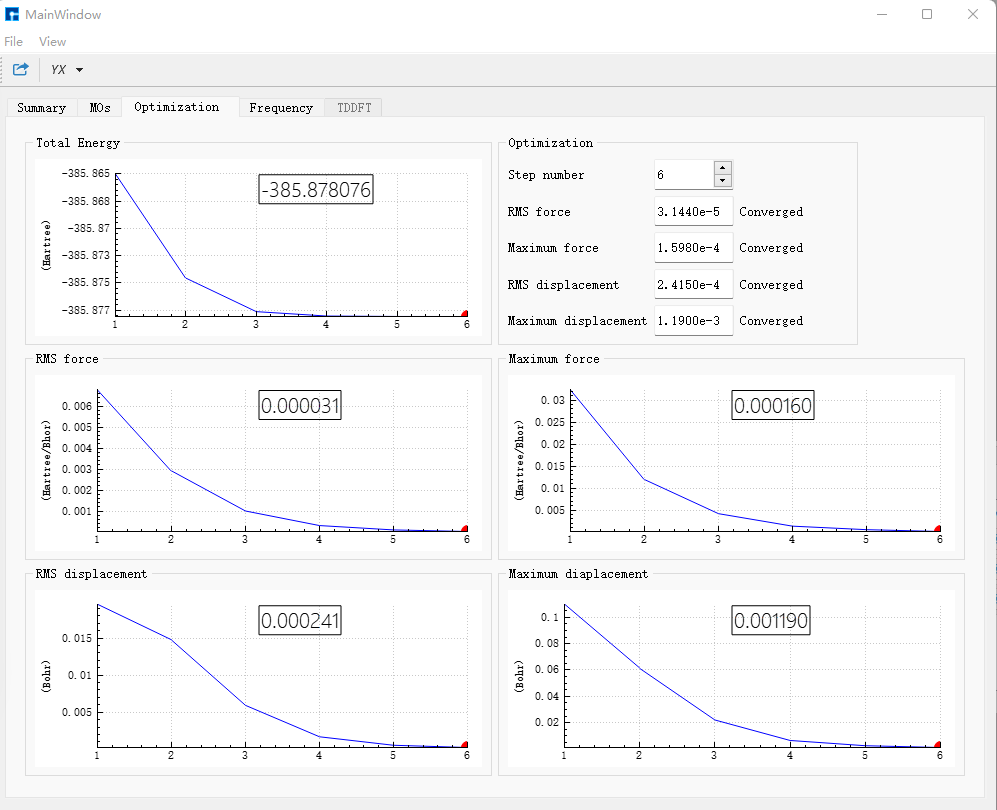
选择bdf.out,右击show view,在Optimization对话框中,显示结构已经达到收敛标准。
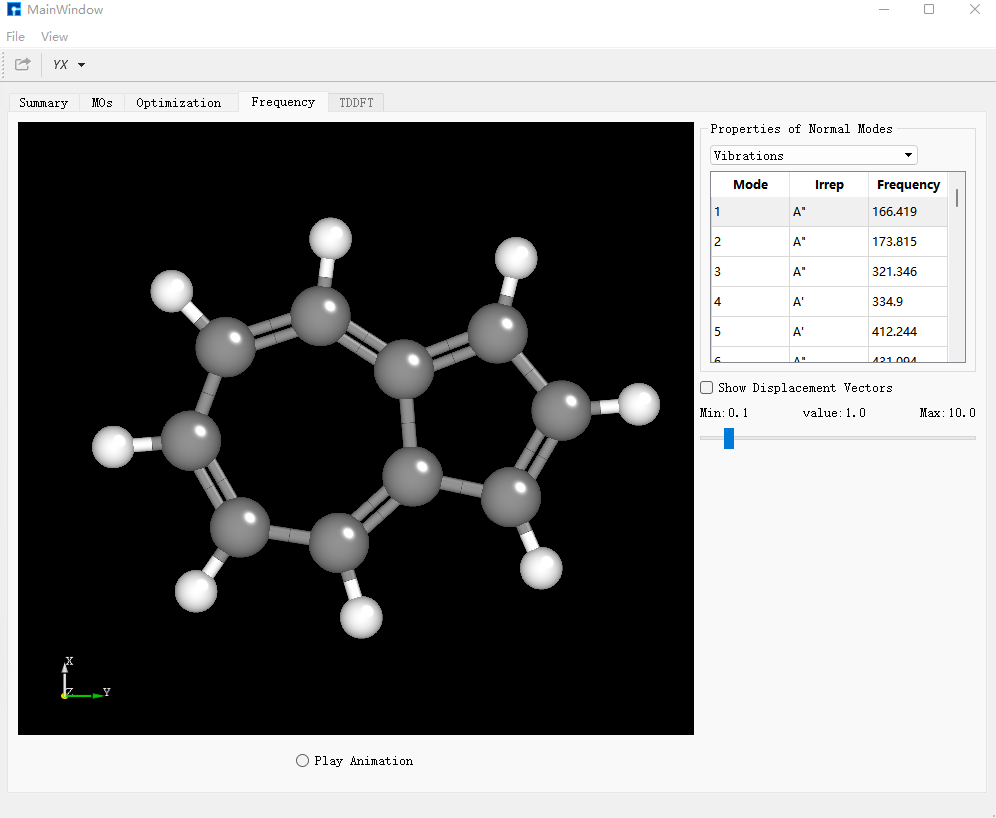
在Frequency对话框中,检查频率,若不存在虚频证明结构已经优化到极小点。
选择bdf.out.tmp,右击open with containing folder,打开bdf.out.tmp,在文件末尾向上查找第一个scf计算模块‘module scf’。该scf模块的Final scf result中的E_tot = -385.87807598为需要的基态azulene的单点能,单位为a.u.。

点击Job Manager中该计算任务,点击服务器,已经进入到了该任务所在文件夹下,输入 /data/hzwtech/DS-BDF_2022A/sbin/optgeom2xyz.py bdf.optgeom,回车,生成bdf.xyz文件。点击文件传输工具,进入文件夹下,将bdf.xyz文件拖出,即为下一步激发态结构优化需要的输入文件。改名为azulene_s0.xyz,打开文件夹,将第二行描述行去掉,拖入Device Studio中。 下一期会接着介绍使用BDF进行azulene的S1激发态结构优化和频率计算和S0→S1之间的非绝热耦合计算。
-
软件
+关注
关注
69文章
5386浏览量
92039 -
辐射
+关注
关注
1文章
611浏览量
38163 -
文件
+关注
关注
1文章
599浏览量
26160
原文标题:鸿之微量化软件计算赏析|量化理论计算探究薁(azulene)的反Kasha规则荧光机制(一)
文章出处:【微信号:hzwtech,微信公众号:鸿之微】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
存内计算技术工具链——量化篇
请问Quartus Ⅱ中怎么把.vhd文件加到.bdf文件(.bdf文件放在顶层)中?
关于qutartus中bdf文件的问题,求解!
Quartus II 9.0 Bdf 无法打开
基于BDF温度场计算优化
量化交易软件开发数字资产自动化交易软件出售
数字资产智能量化交易软件开发自动交易软件开发
数字资产量化交易软件开发自动化交易系统开发
最新科技量化自动交易机器人已开发,交易所量化对冲交易软件
Device Studio应用实例之BDF(上)
Device Studio应用实例之BDF(下)
如何使用MOMAP软件计算S1→S0的荧光辐射速率

GPU:量化理论计算的新引擎




评论