 催化歧化抑制Li-S袋状细胞中的多硫化物穿梭:超越吸附相互作用
催化歧化抑制Li-S袋状细胞中的多硫化物穿梭:超越吸附相互作用
一、全文概要
锂硫(Li-S)电池因其高能量密度和低成本被认为是新一代储能系统的发展方向,近年来受到广泛关注。然而,锂硫电池的商业应用遇到了巨大的障碍,即众所周知的“穿梭效应”现象。在工作条件下,多硫化锂(LiPSs)溶解在电解液中,并在正极和负极之间穿梭,导致库仑效率(CE)低,负极腐蚀严重,因此循环稳定性差。根据已有报道,关于缓解Li-S电池中多硫化物“穿梭效应”的研究,主要集中在导电骨架和正极硫吸附剂的开发。结合高效的负极保护层设计,可以获得具有长循环寿命(2000次循环)和高比容量的实验室扣式电池,或者将多种吸附剂或催化剂复合在正极或隔膜中,以通过非均相物理限制、化学吸附或催化转化来减轻多硫化物的溶解。然而,与实验室扣式电池相比,数百倍甚至数千倍放大的LiPS溶解和穿梭仍然阻碍了高硫含量和贫电解液的Li-S软包电池的商业化。因此,对于实际的Li-S软包电池,需要更有效的分子级均相催化剂来抑制LiPS扩散并提高能量密度。为了解决上述问题,中山大学孟跃中教授和王拴紧教授课题组提出了可溶性添加剂二氟磷酸锂(LiPO2F2)在电解质中可以作为均相催化剂,少量的1wt% LiPO2F2就可以促进LiPS的歧化,以减缓多硫化物的扩散,电解液中含有LiPO2F2的Li-S软包电池具有破纪录的两个月搁置稳定性,长循环后容量保持率从37.0%显著提高到81.4%,并且能量密度超过400 Wh kg-1。此外,不同于已报道的用于减轻LiPS溶解和穿梭的物理/化学吸附或多相催化机制,该研究基于密度泛函理论(DFT)计算首次提出了一种双自由基歧化机制来解释LiPO2F2的催化作用,进一步证明了实验现象和电池性能的改善。该研究以题目为“Catalytic Disproportionation for Suppressing Polysulfide Shuttle in Li-S Pouch Cells: Beyond Adsorption Interactions“的论文发表在材料领域国际顶级期刊《Advanced Energy Materials》。

二、研究亮点
1. 首次在Li-S电池电解质中加入了一种均相催化剂LiPO2F2,以促进LiPSs在S/C正极上歧化,减缓了多硫化物的扩散,减少了活性物质的损失,提升了Li-S电池的循环稳定性。
2. 通过实验和理论计算分析了LiPO2F2的催化机理,证明其是通过双自由基中间体的引发、转移和终止来催化多硫化物歧化。这一对电解质均相催化机理的新认识将为高能量密度Li-S电池的商业化提供新的策略。
三、正文导读
3.1抑制Li-S软包电池中的LiPS溶解
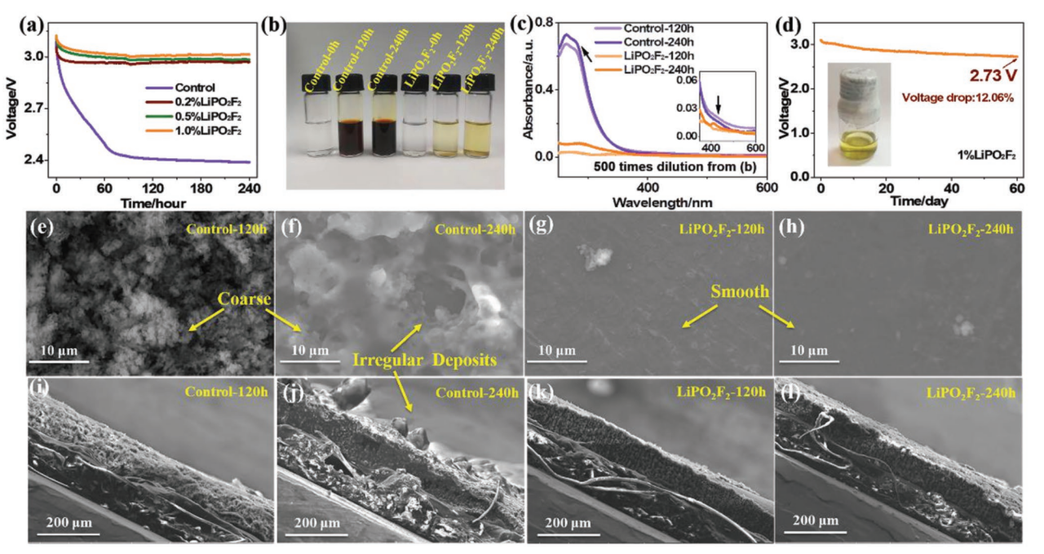
【图1】a)使用含0%(对照电解质)、0.2%、0.5%和1.0%LiPO2F2电解质的Li-S软包电池在240 h静置期间的V-t曲线。b)120 h和240静置试验后软包电池的电解质照片比较。c)从(b)稀释500倍的电解质的紫外-可见吸收光谱。d)添加1%LiPO2F2后静置60天的V-t曲线和插图中电解质的光学照片。在120/240 h静置测试后,软包电池的Li负极的表面e-h)和横截面SEM图像i-l)。
为了组装Li-S软包电池,在电极被电解液完全浸湿后,在第二次密封过程中,要将多余的电解液和气体抽出。然而,电极润湿过程中伴随着严重的锂溶解到电解质中,导致活性S损失和Li腐蚀。在该研究中,使用催化LiPS歧化的LiPO2F2作为电解质添加剂作抑制LiPS的扩散。图1a显示了开路电压(OCV)的变化与含有不同量LiPO2F2的Li-S软包电池在脱气和二次密封之前的静置时间的关系。在具有对照电解质的电池中可以发现明显的OCV下降。40 h后,OCV值迅速下降到约2.4 V,并缓慢下降到Li-S电池的第一放电平台。但是,具有LiPO2F2电解质的电池在测试过程中显示出非常轻微的OCV降低。添加1% LiPO2F2的电池在240 h后仅显示3.51%的最低OCV衰减。在不同的静置时间后,从不同的软包电池中取出多余的电解质,其光学照片如图1b所示,其中较深的颜色代表溶液中LiPS的浓度更高。显然,在试验过程中,对照电解质中产生了大量的LiPS,而即使在240 h的静置时间后,含有LiPO2F2的电解质中只有微量的LiPS,显示出明显的抑制LiPS溶解的作用。它们的UV-vis吸收光谱(图1c)显示了相应的定量结果,表明即使仅在静息120 h后,在对照电解质中存在大量的多硫化物离子(如S42-,S62-,S82-)。相比之下,在120或240 h静置后,在含有LiPO2F2电解质中观察到LiPS的特征吸收可以忽略不计。图1d显示了具有1% LiPO2F2的全Li-S软包电池的OCV曲线,显示了在静置60天后OCV保持2.73 V的高值,仅降低12.06%。图1d中的插图显示,来自相应软包电池的工作电解质在静置60天后仍然表现出非常浅的颜色。
LiPO2F2对LiPS和锂金属之间的副反应的抑制作用可以通过从Li-S软包电池中拆卸下来的锂负极的表面和横截面图像进一步证实。如图1e-l所示,对照电池的锂负极表面粗糙,有大量沉积物,表明有明显的腐蚀。相反,含有LiPO2F2电池的锂负极表面光滑、干净。相应的横截面图像显示锂负极与新负极一样均匀(补充图S1)。XPS显示,在静置测试后,Li2S2/Li2S在对照电池的锂负极上的腐蚀产物比在含有LiPO2F2的电池多得多(补充图S2)。同时,在含有LiPO2F2电池的锂负极上也检测到了LiPO2F2衍生的SEI成分,这表明LiPO2F2在锂表面不可避免地消耗。与LiNO3衍生的稳定界面相比,LiPO2F2衍生的SEI组分未能保护Li负极免受LiPSs腐蚀,这通过浸渍试验得到证明。在Li-Li对称电池中,LiPO2F2对负极界面的保护比LiNO3弱得多,说明负极保护机制并不是抑制LiPS溶解的主要机制。
3.2 改进的Li-S软包电池的电化学性能
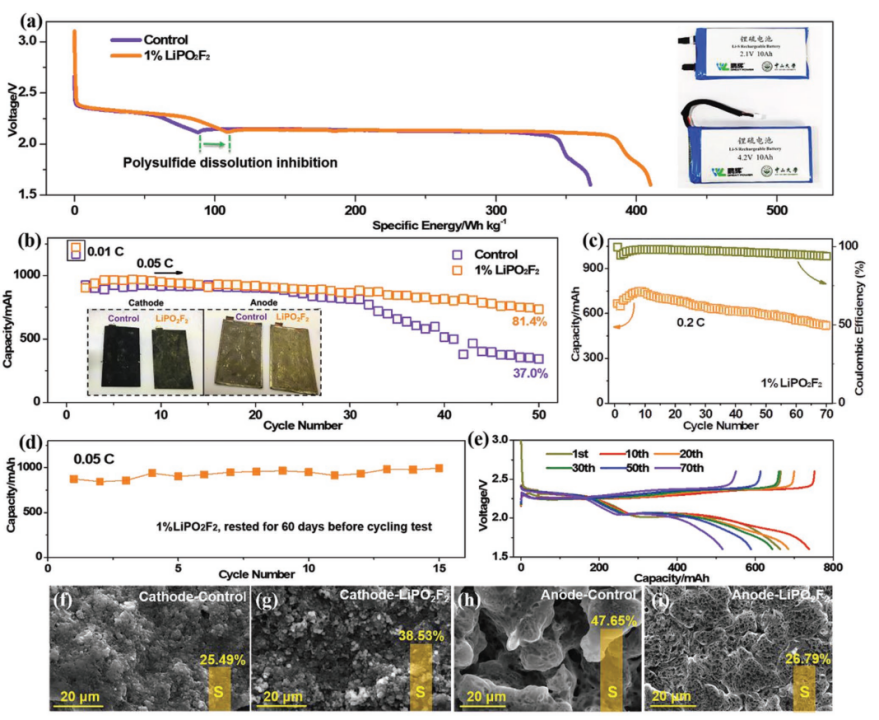
【图2】a)基于总电池在80 mA放电电流下的Li-S软包电池的重量能量密度(5 mg cm-2面积硫负载)。插图:Li-S软包电池的照片。Li-S软包电池(3 mg cm-2面积硫负载)在b)0.05 C下的循环性能,插图:循环后正极和负极的光学照片,c)0.2 C(循环稳定性)和e)相应的充电/放电曲线以及d)60天保存时间后在0.05 C下循环性能,Li-S软包电池在0.05 C下循环50次后的S/C正极和Li负极的照片(b插图)。Li-S软包电池在0.05 C下循环50次后f,g)S/C正极和h,i)负极的SEM图像,对照电池(f,h)和含有LiPO2F2的电池(g,i)。
组装具有5 mg cm-2高面积硫负载的Li-S软包电池,以评估LiPO2F2对能量密度的影响。如图2a所示,含有LiPO2F2的电池比对照电池(367 Wh kg-1)提供更高的质量能量密度(410 Wh kg-1)。增加的能量来源于LiPO2F2对LiPS溶解抑制和反应动力学加速。加快的反应动力学可以通过LiPO2F2电池的CV曲线中更大得电流和较小的过电位来说明(补充图S4)。电压曲线与容量和比容量的关系清楚地表明了添加LiPO2F2后电池容量的改善(补充图S5)。为了了解LiPO2F2添加剂对循环稳定性的影响,在0.05 C(1 C=1675 mA g-1)下测试了面积硫负载为3 mg cm-2的Li/S软包电池。与对照电池相比,使用最佳含量1% LiPO2F2的Li/S软包电池的容量保持率显著提高(从37.0%到81.4%)(图2b和补充图S6a)。在0.2 C下的循环性能和选定的放电/充电曲线如图2c、e所示。含有LiPO2F2的电池在0.2 C时具有740 mAh g-1的高比容量,70次循环后容量保持率为77.8%(补充图S6b)。高于96.6%的平均库仑效率归因于LiPO2F2电池的低自放电率以及LiPO2F2对LiPS穿梭的抑制。与对照电池相比,LiPO2F2电池在0.02至0.3 C的不同电流密度下实现了更高的容量(补充图S7)。在扣式电池中,在0.5 C下循环400次后添加LiPO2F2后容量提高到561 mAh g-1(补充图S8)。图2d显示了在电解液中加入1% LiPO2F2的软包电池在静置60天后的循环性能,结果表明LiPO2F2电池在0.05 C时仍能提供约1000 mAh的高稳定容量,接近静置前的值。
在0.05 C下50次循环后拆卸电池,其正极和负极的外观如图2b的插图所示。LiPO2F2电池的硫正极是黄色的,因为表面上有更多的元素硫,而LiPO2F2电池的锂负极显示出比对照更亮的金属光泽。这表明S已被LiPO2F2添加剂限制在硫正极侧,导致Li负极的腐蚀较轻。对于S/C正极,含有LiPO2F2的电池在第1个循环期间固体硫的空间分布与对照电池不同(补充图S9),由于较高的S利用率,因此对于后续循环更好。经过50次循环后,可以在对照电池的S/C正极上观察到粗糙的表面,在LiPO2F2电池的正极表面上存在蓬松的产物(确认为S)(图2f,g)。LiPO2F2电池的正极表面硫含量为38.53 wt%,远高于对照电池的硫含量(25.49 wt %),表明活性硫的损失受到了限制。该研究推断LiPO2F2与LiPS反应并在正极表面上形成一些硫物质,从而干扰了导电碳的检测。如图2h,i所示,循环后的对照电池的Li负极由于严重的LiPS穿梭而被严重腐蚀,并且在其表面可以发现大量的含S物质(超过47 wt%的元素比率),如Cu元素的存在所示,甚至集电器也暴露了(补充图S10)。相比之下,对于LiPO2F2电池观察到平坦且致密的负极表面,并且S元素仅占26.79 wt%,并且未检测到Cu元素。即使经过20个循环,LiPO2F2电池在96 h的滞留时间内仍表现出较高的OCV(补充图S11)。通过以上讨论,该研究认为Li-S软包电池电化学性能的提高可归因于通过在电解质中添加LiPO2F2抑制LiPS溶解和增强电极动力学。
3.3. 正极上LiPS的歧化
用相同的硫正极和锂负极组装了两个可视的V型电池,分别命名为V-对照电池和V-LiPO2F2电池(补充图S12)。在通过点亮LED灯放电5分钟后,V型电池保持开路状态80分钟。同步测试两个电池,并通过录像机监控(补充视频S1和补充图S12)。在测试过程中,发现添加剂LiPO2F2可以将溶解的LiPSs“带”回硫正极,并抑制它们向锂负极的穿梭。
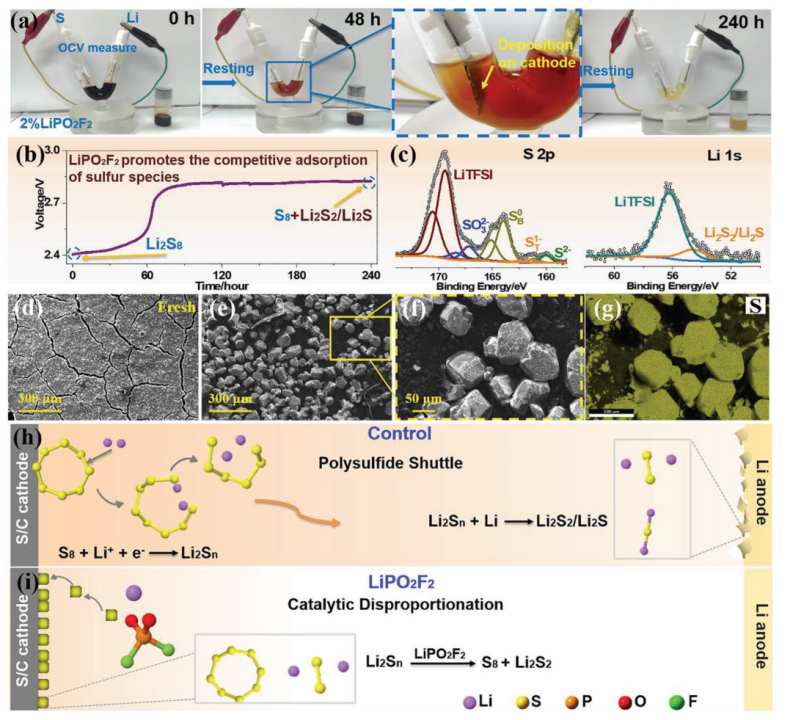
【图3】a)添加2% LiPO2F2后,静置V-型Li/S电池的变化,连同带有含LiPS电解质的玻璃瓶(图1b中的“Control-240 h”)。b)V-型Li/S电池的相应OCV曲线与时间的关系。c)(a)中玻璃瓶沉淀产物的XPS分析。d)新的和f)测试过的eV-型Li-S电池的S/C正极的SEM图像,以及g)相应的S元素映射。h)对照和i)具有LiPO2F2电解质的Li-S电池中LiPS的示意图。
为了进一步研究LiPO2F2对LiPSs的电化学传输的影响,在静置240 h后取自对照电池的电解质(与图1b中的“Control-240 h”相同)用于组装V型Li-S电池。这种电解液含有大量的LiPS,呈现出非常深的棕色。在电解液中加入2% LiPO2F2后,记录电池的OCV并监测外观240 h(图3a和补充图S13)。还同步监测的瓶中含2% LiPO2F2的电解液进行比较。可以看出电解液颜色明显变淡,正极表面被一层新形成的固体沉淀物覆盖。静置240 h后,电解液颜色很浅,代表LiPSs浓度很低,正极表面和V型电池底部有更多的沉积物,为S8和Li2S2/Li2S,这是由于随后LiPS的歧化。电池的OCV值大约从2.4 V开始,这与正极集流体周围的主要Li2S8活性材料相对应(图3b)。电压在开始时缓慢上升,但在60 h后迅速超过2.8 V,表明由于竞争性吸收,主要氧化还原对的跃变在正极中进行。伴随着Li2Sn的歧化,正极中产生了S8和Li2S2/Li2S。形成的具有高氧化态的S8被正极竞争吸附并挤出可溶性LiPSs,导致电极电位升高。这种看似“自充电”的过程在自发歧化反应的帮助下自发发生,其中总吉布斯自由能不可避免地降低。在使用铂片作为工作电极的V型电池中发现了相同的行为,排除S/C材料的影响(补充图S14)。显然,LiPO2F2的电解质添加剂可作为催化剂加速LiPS的化学转化。并且可以通过增加催化剂用量来增强催化效果(补充图S15)。值得注意的是,LiPO2F2催化剂本身在催化过程中没有变化,如19F和31P NMR光谱所示(补充图S16)。
为了证明如上所述由LiPO2F2引发的歧化反应,对歧化产物进行如下验证。经过240 h的静置试验,取出V型Li-S电池中的S/C工作电极,并用DME冲洗以除去可溶性化合物。然后,通过SEM和元素映射分析了电极表面的沉积物。如图3d-g所示,与新鲜的正极相比,观察到了大量具有源自溶解的LiPS的硫物种的块状颗粒。瓶中电解质的颜色(图3a)也缓慢褪色,并在底部形成固体沉淀物,表明LiPSs的歧化反应也可以由LiPO2F2有效触发。值得注意的是,与具有电压差的V型电池相比,瓶子的电解质颜色褪色得更慢。为了进一步确认反应产物,对瓶中的沉淀物进行了分析,以避免来自V型电池的S/C电极的干扰。XPS中的Li 1s和S 2p光谱(图3c)和7Li MAS NMR光谱(补充图S17)表明,LiPS已歧化为不溶性元素硫及其最终还原产物Li2S2/Li2S。在图3c中观察到S 2p由S 2p3/2/2p1/2自旋轨道(ΔE= 1.16 eV;强度比 = 0.511)分量组成的双峰。为了简单起见,接下来将仅讨论2p3/2信号。在宽硫化物区域下的S 2p光谱的反卷积在160.2、161.4和164 eV处显示出三个特征峰,分别对应于Li2S,以及多硫化锂(Li2Sx,x>1)的末端硫(ST1-)和桥接硫(SB0)。硫化物组分的检测归因于电解质中的歧化反应。除了还原产物Li2S2/Li2S(在Li 1s光谱中也检测到了54.6 eV)外,SB0/ ST1-≈ 4.43甚至高于可能更长的多硫化物链(即Li2S8,其中SB0/ ST1-比为3),表明约164 eV的结合能对应于元素硫而不是多硫化物的桥接硫。通过溶解-静置实验研究了添加催化剂后对上述电解质变色和沉淀产生的抑制LiPS溶解的机理,即由于加入LiPO2F2后对LiPSs歧化反应的作用催化而不是多硫化物溶解度的降低(补充图S18)。
如图3h,i所示,在催化歧化过程中,LiPO2F2催化剂不断将溶解的LiPSs带回正极,从而保护锂负极。对于图1中的实际Li-S软包电池的静置,由于可溶性S8和Li的反应,连续的LIPSs溶解到电解质中,导致电池电压和容量降低。因此,电解质中溶解的LIPSs将在形成后的脱气过程中被除去,或者在电解质或隔膜中自发地歧化。加入LiPO2F2后,电解液中不可避免产生的LIPSs被选择性地“携带”到正极,歧化为不溶性的S8和Li2S2/Li2S,抑制了可溶性LIPSs的进一步生成。在静置过程中,LiPO2F2的加入引发了自发的LIPS歧化,并抑制了S在电解质中的严重溶解。此外,在脱气和二次密封过程中,有效地减少了活性LIPSs的损失。显然,在LIPSs的初始自发歧化过程中,通过牺牲少量的吉布斯自由能,将更多的活性硫物种保留在电池中用于循环测试,从而提高了比容量和能量密度。
3.4 计算通过双自由基的歧化途径
在报道的吸附催化模型中,通常通过计算催化剂与LiPS之间的吸附能来评价催化行为,这与实验中报道的LiPS吸附机理一致。然而,在该研究的催化歧化模型中,提出了歧化的起源和通过双自由基的歧化路线。
以LiPS的Li2S8为例(Li2S6和Li2S4也可以歧化,Li2S8是初始放电产物),当S的氧化态高度一致时,Li2S8会稳定,但随着添加的催化剂改变氧了化态,Li2S8将发生歧化(例如,LiPS歧化可由水溶液中的H+触发,等式(1))。

在非水溶剂体系中,Li2S8的氧化态被认为受锂盐的影响。为了揭示不同添加剂和Li2Sn结合的复杂分子的氧化还原特性,分析了其前沿分子轨道(FMOs)理论的最低未占据分子轨道(LUMO)和最高占据分子轨道(HOMO)的能级。如图4a和补充表S3所示,与不同锂盐的组合确实改变了Li2S8的能级。与Li2S8和其他催化剂-Li2S8配合物相比,LiPO2F2-Li2S8和LiBOB-Li2S8的LUMO值显著降低(超过0.2 eV),而HOMO值没有太大变化,这意味着缺电子催化作用与Li2Sn结合可以得到高度氧化的LiPO2F2-Li2Sn或LiBOB-Li2Sn。在该研究的实验中,在电解质中形成少量的强氧化剂(例如LiPO2F2-Li2Sn和LiBOB-Li2Sn)氧化LiPS底物被认为是歧化现象的起源(等式(2))。
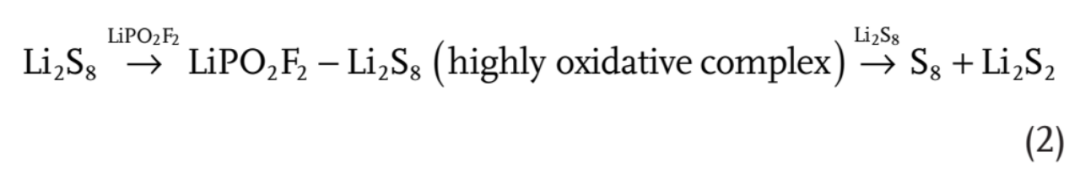
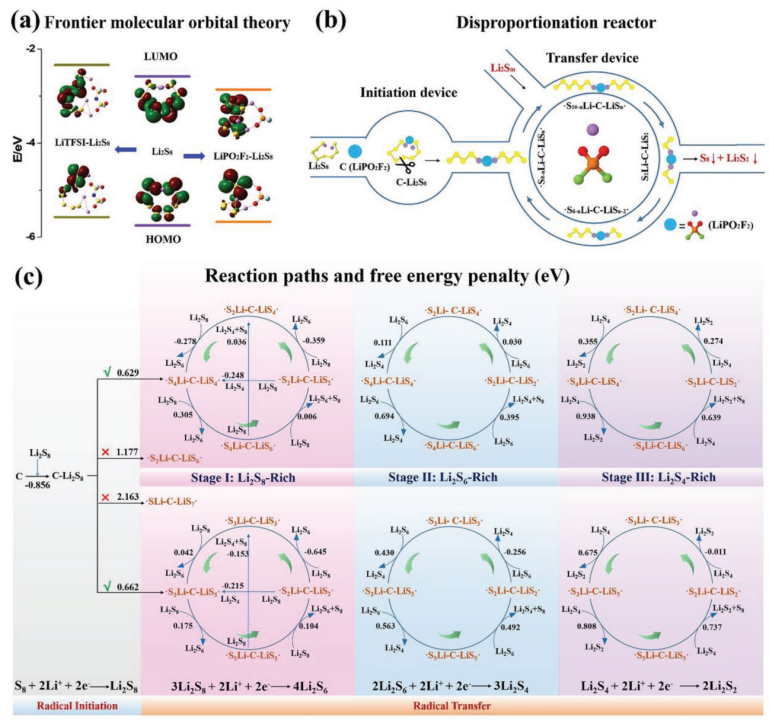
【图4】a)Li2S8、LiTFSI-Li2S8和LiPO2F2-Li2S8的前沿轨道对应的真空能量。b)示意图和c)双自由基在LiPO2F2在Li-S电池放电或自放电下多硫化物歧化中可能的反应路径。
在Li-S电池放电机理的研究中,一个被广泛接受的观点是自由基的存在,并可能在Li-S电池中发挥重要作用。利用DFT计算中的不同泛函集和基集已经被用来研究碱金属-S电池中的各种多硫化物单自由基,特别是S3․-。因此用DFT计算方法研究来研究通过双自由基进行歧化的独特路径。
在没有LiPO2F2催化剂的情况下,从Li2S8引发自由基LiS4·+LiS4·或LiS3·+LiS5·以及从Li2S6引发自由基LiS3·+LiS3·分别需要0.831、0.985和0.746 eV的能量。在LiPO2F2催化剂存在的情况下,单自由基C-LiS4·+LiS4·(C,催化剂,代表LiPO2F2)、C-LiS3·+LiS5·和C-LiS3·+LIS3·的引发能量分别为0.932、1.107和0.849 eV(补充图S19),均高于不含LiPO2F2催化剂的情况。然而,C-Li2S8可能分裂为双自由基·S4Li-C-LiS4·或·S3Li-C-LiS5·,而C-Li2S6则产生·S3Li-C-LiS3·,其自由能损失分别降至0.629、0.662和0.601 eV。这是因为LiPS更易于形成连接在LiPO2F2催化剂上的环状结构(补充图S20),其中S8链可能容易断裂并形成双自由基。这表明LiPO2F2通过双自由基中间体加速了LIPS的歧化。
给出了LiPS、LiPO2F2、多硫化物LiPO2F2配位配合物、单自由基和双自由基(单线态和三线态)的优化结构,以及它们在溶剂化和非溶剂化模型中相应的能量和电子自旋密度(补充图S21-S25和补充表S4)。该文还给出了溶剂化模型中基于三重态的结果。计算了通过单自由基的LiPS歧化反应的能量分布,并列出了在有催化剂和没有催化剂的情况下LiPS与单自由基之间所有可能的反应路径(补充图S26和补充表S5-S6)。如图4b所示,提出了双自由基机理用于解释LiPS的歧化:催化剂诱导双自由基的产生,LiPS的歧化反应是通过双自由基之间的自由基转移来实现的。
具体反应路径如图4c所示,在放电或自放电开始时,Li2S8在S电极处通过收集电子和Li+产生。在LiPO2F2的帮助下,Li2S8很容易转变为·S4Li–C–LiS4·或·S3Li–C–LiS5·。通过链转移反应,这些双自由基将硫碎片(Sx)转移到LiPSs上并形成更高阶的LiPSs,直到形成不溶的S8(如·S4Li-C-LiS6· + Li2S8= ·S2Li-C-LiS2· + Li2S6+ S8)。它们还可以从高阶LiPSs中脱去硫碎片,并将它们转化为低阶LiPSs,直到不溶解Li2S2(如·S2Li-C-LiS2·+Li2S4= ·S2Li-C-LiS4·+Li2S2)。这两个链转移反应将不断消耗LiPS,并将其转化为无法溶解的S8和Li2S2。值得注意的是,自由基反应伴随着电子转移进行,因此锂硫电池中的自放电会促进自由基反应动力学。因此,由于电压偏压的存在,V型电池中的 LiPS歧化速率比瓶中更快(图3a)。
根据放电深度的不同,双自由基转移反应可以分为三个阶段,其中Li2S8、Li2S6和Li2S4分别是消耗的反应物(补充图S27)。在包括两个Li2Sn分子的歧化反应的可能的反应路径中,来自两个Li2S8的能量最高的有利反应路径需要增加0.696 eV的自由能(补充表S7和补充图S28)。在该反应中,能量上最有利的反应路径是通过双自由基实现的,速率控制步骤仅为0.395 eV。事实上,形成固体产物的能量低于计算的沉淀分子的能量(补充图S29)。因此,这些反应的自由能损失也应该更低。此外,还比较了富Li2S8(阶段I)、富Li2S6(阶段II)和富Li2S4(阶段III)环境中自由基引发和转移的可能反应路径,以了解其可行性(补充图S30)。
从第一阶段到第三阶段,自由基转移的能量逐渐增加。由于LIPSs和LiPO2F2的强结合能,键合的LIPSs不能转变为S8和Li2S2沉淀。当未键合的LIPSs浓度降低甚至全部消耗时,高度不稳定的双自由基的碰撞概率增加,歧化反应仍然发生(补充图S31)。伴随着双自由基的不断消耗,其反应概率会将在一定程度上降低,并且双自由基·SxLi-C-Lisn-x·的踪迹将转变为C-Li2Sn,并变得动态平衡(补充图S32)。因此,从自由基引发到终止,Li2Sn歧化需要六步反应路径。在自由基转移循环期间,初始反应物决定该循环的起始点(补充表S8)。
为了验证所提出的催化过程中双自由基形成的机理,除了LiPO2F2,还通过对Li-S软包电池进行电压监测,探索了LiFSI、LiBOB、LiClO4、LiPF6等其他商业锂盐在锂电池中的催化效果,并计算了相应的双自由基引发能(补充图S33-S35)。实验结果与计算结果的一致性表明,LiBOB、LiPO2F2和LiPF6等锂盐添加剂作为有效的催化剂,可以促进LiPS歧化反应通过双自由基途径进行。因此,上述想法将为优化催化剂/电解质以实现锂硫电池的商业化提供指导。
3.5 自由基特征的实验验证
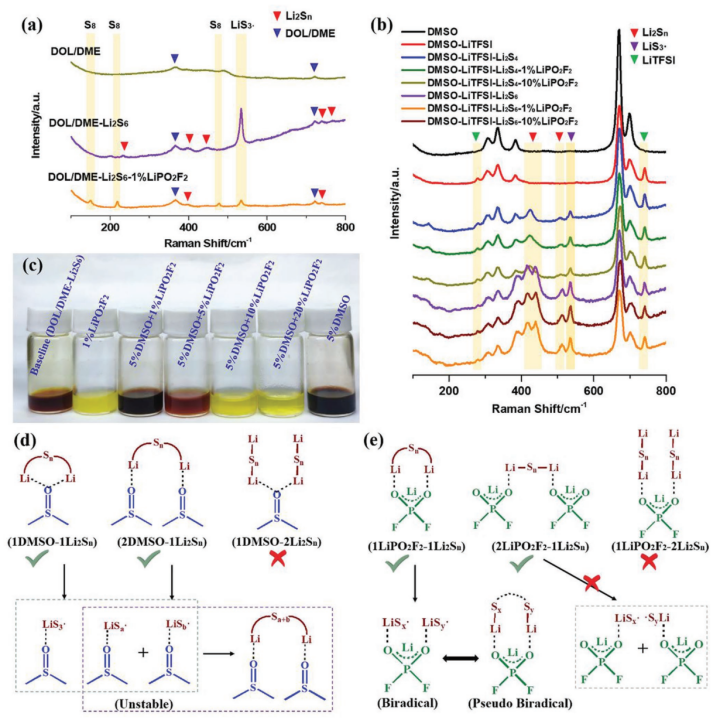
【图5】a)DOL/DME、DOL/DME-Li2S6和DOL/DME-Li2S6-1% LiPO2F2,b)加入不同量LiPO2F2的DMSO-Li2S4和DMSO-Li2S6溶液的拉曼光谱,其中使用1 M LiTFSI作为内部标记物和纯DMSO。c)(DOL/DME-Li2S6)和240 h后含不同剂量的DMSO和LiPO2F2溶液的光学照片。d)DMSO和e)LiPO2F2与Li2Sn的自由基引发机理。
具有未配对电子的物种在电子顺磁共振(EPR)光谱中会产生强信号。在DOL/DME-Li2Sn溶液体系中,EPR光谱中存在少量自由基信号(补充图S36),表明DOL/DME基电解质中存在微量自由基。然而,由于自由基阴离子在双配位硫原子上的高极化率,拉曼光谱可以很好地检测平衡条件下低浓度的自由基阴离子。如图5a所示,除了Li2Sn外,在DOL/DME-Li2S6溶液的拉曼光谱中,在535 cm-1处出现了明显的不稳定LiS3·自由基信号。添加1% LiPO2F2后,Li2Sn和LiS3·的峰信号降低,在156、221和 473 cm-1处发现了对应于硫的新峰,表明高氧化性LiPO2F2-Li2Sn氧化了LiPS底物,导致LiPS的歧化,从而降低自由基浓度。
由碱金属多硫化物在高供体溶剂中的溶液形成的硫自由基离子Sn-(n = 2-4)已得到充分证明。LiPS在高供体电解质中的高溶解度与LiPS不同的歧化和解离途径有关,这些途径在低供体电解质中是不可用的。DMSO作为电子对供体溶剂用于提高硫自由基阴离子的稳定性。正如预期的那样,自由基信号可以通过EPR在DMSO-Li2S4和DMSO-Li2S6溶液中明显捕获,此外,LiS3·在617 nm处的吸光度也出现在紫外-可见光谱中(补充图S37和S38)。与低给体溶剂DOL/DME相比,DMSO确实稳定了硫自由基。此外,在DMSO-Li2S4和DMSO-Li2S6溶液中加入1%和10% LiPO2F2后均未发现可见沉淀,表明LiPS和LiS3·在DMSO基高给体溶剂中均稳定,这与补充表S3中DMF-Li2S8和DMSO-Li2S8的能级相呼应。然而,当添加LiPO2F2时,EPR中的顺磁信号和自由基紫外吸光度略有下降,说明DMSO稳定了LiS3·,从而抑制了LiPS歧化,而LiPO2F2促进了LiPS歧化,从而导致自由基(单自由基或双自由基)浓度降低,根据等式(3):
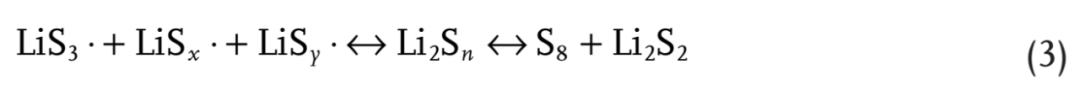
拉曼光谱被用来进一步支持上述假设。在溶液中加入1 M LiTFSI作为内标物质,用于校准其他物种的信号,其信号位于278和741 cm-1。如图5b所示,通过在DMSO-Li2S4或DMSO-Li2S6溶液中加入LiPO2F2,分配给可溶性LiPSs和LiS3·的峰变弱,表示方程式(3)中的平衡移至S8和Li2S2沉淀。由于DMSO分子极大地稳定了自由基,因此DMSO溶剂中LiPS的歧化程度非常低。因此,当加入LiPO2F2时,在拉曼光谱中没有检测到元素S8。在DOL/DME–Li2S6溶液中,LiPO2F2和DMSO分子之间竞争的视觉歧化现象(图5c和补充图S39)进一步证实LiPO2F2与DMSO分别促进和抑制LiPS歧化。
DMSO稳定LiS3·和LiPO2F2促进双自由基的可能反应机理如图5d,e所示。在图5d中,存在三种在Li2Sn和DMSO之间形成Li键的方式(1DMSO-1Li2Sn、2DMSO-Li2Sn和1DMSO-2Li2Sn)。作为较好的锂键受体,DMSO通过1DMSO-1Li2Sn或2DMSO-Li2Sn形成相对稳定的锂键。这两种配合物中的S-S键很容易断裂以生成不同的单自由基,而除LiS3·以外的这些单自由基不稳定,并且倾向于重组为中性分子(2DMSO-Li2Sa+b,a+b = n - 3 ≠ 3)。同样,LiPO2F2的独特特性使其自发形成1LiPO2F2-1Li2Sn和2LiPO2F2-1Li2Sn的两个强Li键,如图5e所示。正如在DFT计算中讨论的那样,根据变化的S-S键长,可以从1LiPO2F2-1Li2Sn形成不同的双自由基。在LiPO2F2-Li2Sn中,根据S-S键长度的不同,可以形成具有典型双基特征的LiPO2F2-LiSx-LiSy(x + y= n)(补充图S40和补充表S9)。与由1LiPO2F2-1Li2Sn形成的双自由基相比,由2LiPO2F2-1Li2Sn形成的LiPO2F2-LiSx·和LiPO2F2-LiSy·单自由基需要更高的能量,这是不可行的路径(例如,0.94 eV的2LiPO2F2-1Li2S8形成2LiPO2F2-LiS4·,0.96 eV的2LiPO2F2-1Li2S6形成2LiPO2F2-LiS3·)。
四、总结和展望
这项工作首次提出了一种可用于在Li-S电池电解质中的均相催化剂LiPO2F2,以促进LiPS在S/C正极上的歧化。歧化产物是S和Li2S2/Li2S。通过实验现象和理论分析详细说明了LiPO2F2添加剂的催化机理,证明其通过双自由基中间体引发、转移和终止来完成。在LiPO2F2的帮助下,自由基引发的能垒显着降低,从而促进了整个歧化过程。快速转化反应降低了可溶性LiPSs的停留时间和Li2S/Li2S2成核的动能势垒,进而减少了活性材料的损失并加速了电极的氧化还原动力学。因此,该研究最终设计并制造了一种Li-S软包电池,其保存期达到创纪录的2个月,循环稳定性显着提高(容量保持率从37.0%提高到81.4%),并且能量密度超过400 Wh kg-1.
审核编辑 :李倩
-
催化剂
+关注
关注
0文章
92浏览量
10314 -
能量密度
+关注
关注
6文章
287浏览量
16502 -
软包电池
+关注
关注
1文章
175浏览量
8007
原文标题:中山大学AEM:Li-S软包电池最新突破,催化歧化超越吸附作用,抑制多硫化物穿梭效应
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
清华深研院刘思捷/港科大Kristiaan Neyts最新AEM封面文章:硫化物复合固态电解质

LED中硫化物的来源及其对性能的影响:以硫为例

离子束与材料的相互作用
OptiSystem应用:EDFA中离子-离子相互作用效应
原位焊接离子导电断点以实现高度可逆的全固态锂硫电池

电子耦合在电路中的作用
华为公布硫化物固态电池新专利,固态电池技术加速发展
孙华军:硫化物固态电池预计2027年起步入示范应用阶段
EthernetiP转modbusTCP网关在加氢催化中的应用

弦采集仪岩石桩基施工相互作用监测中的几个方面

一种基于可拉伸光子晶体的荧光传感阵列,用于卵巢癌早期诊断
将废正极材料升级为高稳定性锂硫电池的双功能催化剂!

用光子连接悬浮在真空中的纳米粒子,并控制它们之间的相互作用
美国研究人员使用干细胞制作芯片心脏,助力药物安全性评估
2023年锂电池研究重大突破






 工商网监
工商网监
评论