 浓度极化诱导相变稳定聚合物电解质中的锂镀
浓度极化诱导相变稳定聚合物电解质中的锂镀
背景介绍
锂金属负极具有3860 mAh g-1的理论容量,以及超低的电极电位(-3.04 V),因此有望极大提高电池能量密度。然而,液体电解质中的浓差极化促进了金属沉积过程中晶须的生长,导致锂沉积不均匀,形成粗糙的形貌,如苔藓状和树枝状的锂。这种不均匀沉积不仅导致电极表面积大,促进与电解质发生副作用,降低库仑效率(CE)和循环寿命,而且还存在内短路、热失控等安全隐患,尤其是与传统的易燃液体电解质(如醚类、碳酸盐)匹配时。聚合物电解质虽然不能完全解决锂金属电池(LMBs)中的安全问题,但由于其比液体电解质的热稳定性更高,因此有助于提高LMBs安全性。遗憾的是,聚乙烯氧化物(PEO)电解质的杨氏模量,通常在20-70 MPa范围内,远低于有效抑制Li晶须所需的1 GPa阈值。因此,Li晶须在聚合物电解质中快速生长。当引入增塑剂来增强离子电导率时,会进一步软化聚合物电解质,晶须生长变得更加严重。解决这一挑战需要对动态的Li金属/聚合物电解质界面有基本的理解,例如Li+浓度如何在Li负极表面演变,以及Li负极如何与固体电解质相互作用。
二、正文部分
1、成果简介
近日,哥伦比亚大学杨远教授,闵玮教授和Qian Cheng,通过受激拉曼散射显微镜证明,固体聚合物电解质中的浓差极化可以诱导聚乙烯氧化物(PEO)电解质发生相变,在锂/电解质界面形成一个富PEO但贫盐/增塑剂的新相。与本体聚合物电解质(《1 MPa)相比,新相具有高得多的杨氏模量(~1-3 GPa)。因此,PEO电解质的组成应在相图中单相区和两相区的边界附近,这样施加的电流可以诱导形成机械刚性的富PEO相来抑制锂晶须。具有浓差极化诱导相变的LiFePO4/PEO/Li电池可以可逆循环100次,而没有这种相变的电池在10次循环内失效,证明了该策略的有效性。该研究以题目为“Stabilizing lithium plating in polymer electrolytes by concentration-polarization-induced phase transformation”的论文发表在国际顶级期刊《Joule》上。

2、研究亮点
本工作利用具有高时间分辨率、成像速度和灵敏度的受激拉曼散射(SRS)显微镜研究了固体聚合物电解质(SPE)与电极的相互作用。结果表明,浓差极化并没有促进晶须的生成,而是降低了锂/电解质界面的盐浓度,使单相PEO电解质转变为两相PEO电解质。这导致在锂/电解质界面形成了模量为~1-3 GPa的机械刚性富PEO相,对应的剪切模量为0.36-1.06 GPa(图1B)。这么的高模量抑制了晶须的生长,使锂沉积均匀。因此,电解质成分应位于PEO-盐-增塑剂相图中单相区和两相区的边界处,这样通过小电流诱导形成轻微的浓差极化,就可以在金属锂表面获得机械刚性的富PEO相,使金属锂钝化。具有浓差极化诱导机制的LiFePO4(LFP)/PEO/Li电池能够稳定循环超过100次。
3、图文导读
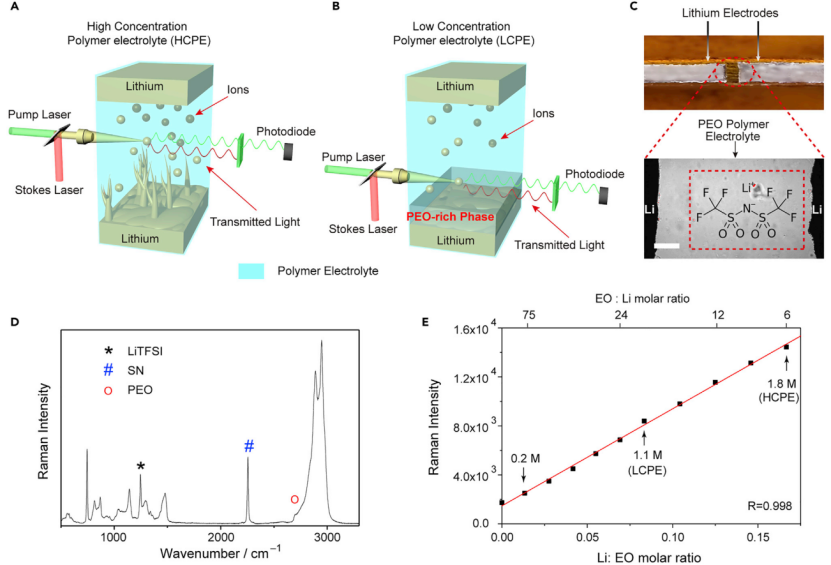
【图1】(A和B)Li/Li电池中高浓度聚合物电解质(HCPE)和低浓度聚合物电解质(LCPE)的SRS成像示意图。(C)Li/PEO/Li电池的亮场图像。上图为电池结构,下图为光学显微镜放大后的图像,比例尺为100 mm。(D)LCPE的拉曼光谱。LCPE对应的组成为EOSN=122.64(摩尔浓度)。(E)1245 cm-1处LiTFSI峰的拉曼强度图随PEO电解质中Li:EO比的演变,显示出良好的线性关系。1.1 M和1.8 M LiTFSI的点分别对应LCPE和HCPE。
自制了锂对称电池,并通过SRS显微镜研究Li/PEO电解质相互作用(图1C)。在该电池中,PEO电解液填充在两块锂之间的空隙中,所有组件被夹在两块玻片之间,用环氧树脂密封。PEO电解质以双(三氟甲磺酰)亚胺锂(LiTFSI)为盐,琥珀腈(SN)为增塑剂,以增强离子电导率,使其可在室温(RT)下运行。图1D的LCPE拉曼光谱显示,在1245 cm-1(C-F拉伸)、2250 cm-1(C≡N拉伸)和2800 cm-1(C-H的拉伸振动)附近的峰分别为LiTFSI、SN和PEO的拉曼特征峰(图1D)。由于电解质满足电中性要求,因此[Li+]可以被认为等于[TFSI-]。测量[TFSI-]可以用来表示局部的[Li+]。而TFSI-的拉曼强度与其浓度成正比,因此可以将拉曼信号转换为化学浓度(图1E)。
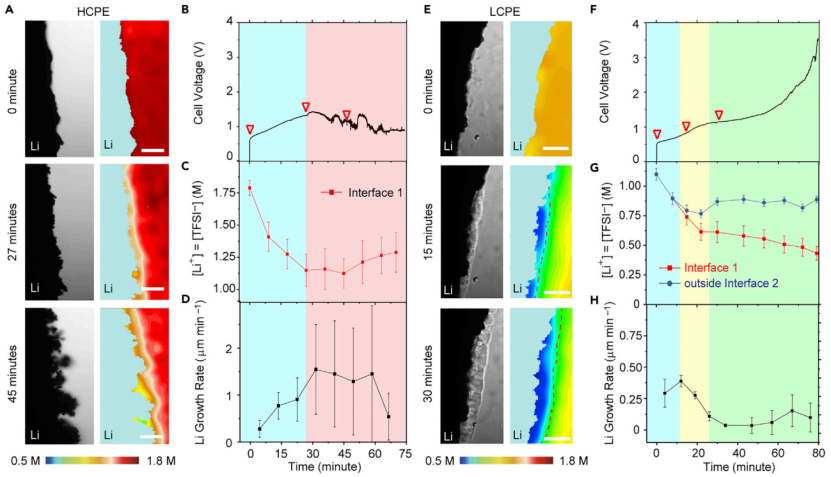
【图2】(A-D)HCPE中锂晶须的生长。(A)[Li+]=[TFSI-]三个代表性阶段的亮场及对应的SRS图像。(B)(A)中Li/Li电池的电压曲线。(C)[Li+]=[TFSI-]随时间的变化,(D)锂生长速率(v)随时间的变化。界面1为锂电极与PEO电解质的边界。(E-H)锂在LCPE中的生长。(E)[Li+]三个代表性阶段的亮场图像和SRS图像。(F)(E)中Li/Li电池的电压曲线。黄色的阴影对应富PEO相的出现,绿色的阴影表示富PEO相已经覆盖整个锂表面。(G)[Li+]=[TFSI-]随时间的变化,(H)锂生长速率(v)随时间的变化。界面2为富PEO相与各向同性体相聚合物电解质之间的边界,如图(E)中SRS图像中的虚线所示。比例尺为50 mm。两个电池的电极距离均为500mm。在室温下,对两个电池施加0.5 mA cm-2的电流密度。
以EOSN=122.64和122.64,分别制备了高浓度聚合物电解质(HCPE)和低浓度聚合物电解质(LCPE),并进行了研究,HCPE对应的LiTFSI和SN浓度分别为1.8 M和2.4 M, LCPE对应的LiTFSI和SN浓度分别为1.1 M和2.9 M。两种情况下SN与PEO的重量比均固定在40%。
对于HCPE(图2A),施加0.5 mA cm-2的电流时,锂表面的[Li+]([Li+]0 μm)从t=0时的1.8 M逐渐消耗到t=27 min时的1.2 M,之后[Li+]0 μm在稳定在~1.2 M(图2B和2C)。同时,在t=0的锂生长速率(v)从0.27±0.18 μm min-1迅速增加到t=27 min的0.9±0.46 μm min-1(图2D)。之后,v在剩余的时间里急剧增加到~1.5 μm min-1,导致了97%的超高孔隙率,表明HCPE不能抑制晶须的生长。这些结果表明,如果不发生相变,Li/聚合物电解质界面的离子浓差极化促进了晶须的生长。
图2E和2F的SRS图像和明场(BF)图像显示,与HCPE中观察到的行为相反,LCPE中的浓差极化导致锂/电解质界面发生相变过程,这反而能抑制锂晶须的生长。首先,在SRS中,新相出现在蓝色区域,在BF中表现为粒状区域。从Li/电解质界面(界面1)上的[LiTFSI]和新相与各向同性体相电解质界面(界面2)外的[LiTFSI]对比可以看出,富PEO相的[LiTFSI]比相邻各向同性体相电解质中的[LiTFSI]低得多(图2G)。这一差异在t=15 min时为0.79/0.74 M,在t=30 min时为0.87/0.61 M,在t=79 min时为0.89/0.43 M。
这一新相的出现能够有效地抑制锂晶须的生长。虽然在t=0时观察到锂晶须生长,v为~0.3 μm min-1(87%孔隙率),但在t=30 min时,富PEO相初步形成后,v迅速下降到0.048 μm min-1(图2H),相当于孔隙率为16%。在这一阶段,富PEO相逐渐在金属锂表面形成。新相完全覆盖Li金属表面后,30 ~ 63 min的平均v仅为0.044 μm min-1,孔隙率为9.2%,约为HCPE的三十分之一,表明锂沉积致密均匀。这种行为在浓差极化过程中不仅是自发的,而且是自增强的。例如,如果锂晶须在某一位置生长迅速,局域电流密度会增加,导致浓差极化更大,从而形成更厚的富PEO相,从而抑制晶须的生长。
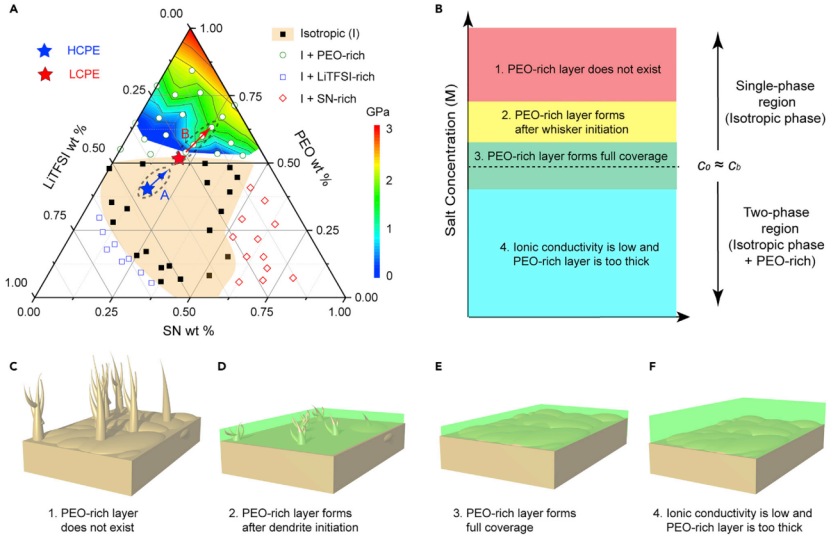
【图3】(A)PEO-LiTFSI-SN三元相图。橙色代表各向同性单相区域,其他区域代表两相区域。三角形顶部的彩虹色区域代表相图中相应组分形成的富PEO相杨氏模量分布。(B)当施加电流时,聚合物电解质中初始盐浓度影响富PEO层厚度和覆盖面积的示意图。(C)当c0较高时,没有形成富PEO层,容易形成锂晶须(c0cb)。(D)c0略高于cb,因此晶须通常比富PEO层更早形成。这导致了锂晶须生长的部分钝化。(E)c0cb时,富PEO相对金属锂的钝化效果最好。(F)c0《cb时,它具有较低的离子电导率,可能导致厚的富PEO层。
在排除电解质分解的可能性后,在高通量SRS的帮助下构建了PEO-LiTFSI-SN三元相图(图3A),以研究相随成分的演化。相图由中间(橙色)的单相各向同性区(I区)和角落处的三个两相区(白色区)组成。图3A显示,HCPE的组成位于I区中心。当施加电流时,锂金属表面的[LiTFSI]逐渐降低(图3A中的路径A)。由于HCPE中盐浓度较高,浓极化过程中Li/电解质界面处的电解质成分保持在I区,没有发生相变。
相比之下,LCPE的组成非常接近单相I区和两相区的边界(cb),容易发生浓差极化(图3A中的路径B和3E),因为形成两相比保持单相在热力学上更稳定。由于较大的离子浓度梯度,较低的t+可以在较早的时间触发相变。这种相变不仅使新相的[LiTFSI]降低,而且使[SN]降低,将SN排斥到各向同性电解质中。
在EOSN=82.64的Li/Li电池中,盐浓度略高于cb。锂沉积开始后,锂晶须先生长,未观察到富PEO相。而当锂表面盐浓度逐渐降低至低于cb时,富PEO相出现并增厚,抑制了锂晶须的生长。但由于锂晶须最初呈针状,导致富PEO相不能完全覆盖在锂表面,破坏了保护的有效性。最后,测试了浓度为0.52 M LiTFSI和2.6 M SN的聚合物电解质,该聚合物电解质位于两相区深处。这种固体电解质具有较低的离子电导率,无法正常运行。
从以上四种情况可以得出结论,富PEO层来源于RT时或近RT时的固相转变,其形成高度依赖于PEO电解质中的盐浓度(图3B)。如果初始盐浓度(c0)在单相区较深处,则HCPE(c0cb)时,不太可能形成富PEO相(图3C)。当c0略高于cb时,晶须可能在富PEO相形成之前就开始形成,因此不能完全抑制晶须生长(图3D)。如果c0接近cb(LCPE),即使很小的电流也能形成富PEO相,在短时间内完全覆盖锂表面,有效抑制晶须(图3E)。如果c0《cb,聚合物电解质的离子电导率通常较低。此外,富PEO的相很容易在金属锂表面形成,但它可能太厚而不能导电(图3F)。
基于以上分析,提出了LMBs聚合物电解质的设计原则:电解质成分应位于PEO-盐-增塑剂相图中单相区域和两相区域的边界处,这样只需要很小的电流就会导致盐浓度降低,并在金属锂表面形成机械强度高的富PEO相。
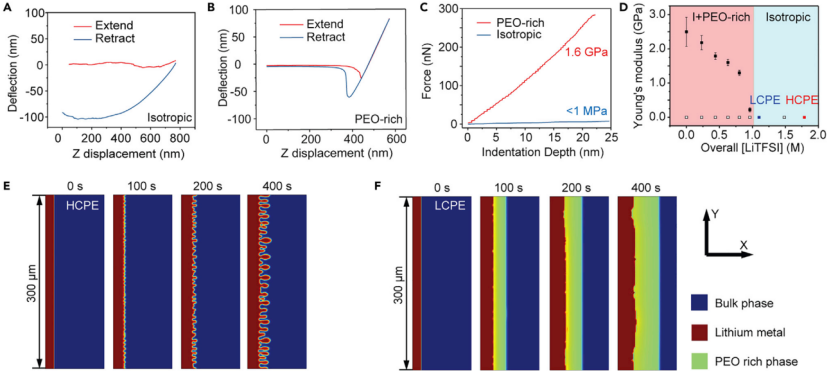
【图4】(A)各向同性体相电解质(I区,HCPE)和(B)富PEO相的AFM拉伸和收缩力曲线。(C)(A)和(B)样品对应的力-压痕曲线。(D)不同盐浓度下富PEO相的杨氏模量。(E)高浓度(2 M盐)和(F)低浓度(1 M盐)固体聚合物电解质在 0.5 mA cm-2下锂电沉积的相场模拟。
富PEO相对锂晶须的有效抑制是由锂/电解质界面相变过程中的机械力-化学耦合引起的。为了验证这一点,使用AFM测量了不同组成的SPE杨氏模量,包括各向同性相(I区)和富PEO相,并将结果叠加在图3A的相图上。对于HCPE(1.8 M LiTFSI, 2.4 M SN),接近曲线显示,当尖端压入电解质时,悬臂梁没有变形,说明电解质非常柔软(图4A)。针尖缩回时检测到大的悬臂挠度,证实针尖被压在HCPE内部,电解质有粘性。图3A中I区内的所有SPE和两相区域中SPE的各向同性相显示出相似的结果(杨氏模量《1 MPa),表明它们不能抑制锂晶须的生长。
相反,SPE中形成的富PEO相表现出不同的力曲线。样品整体组成为0.6 M LiTFSI, 3.3 M SN,其中富PEO相包含0.52 M LiTFSI和2.6 M SN(图4B)。采用Sneddon模型,对应的模量为1.6 GPa(图4C),压痕硬度为~410-780 MPa。材料的压痕硬度通常是其抗拉强度的3倍,这意味着富PEO相的抗拉强度为~137 MPa。这远远超过了金属锂的硬度(~7-43 MPa)和屈服强度(0.6-1.3 MPa),所以在锂沉积过程中金属锂的蠕变是不可避免的,导致锂晶须的生长受到抑制。而体相PEO电解质的模量《1 MPa,与金属锂的硬度和屈服强度相近或较小,因此不能抑制锂晶须的生长。
为了进一步了解Li/电解质界面上的机械力-化学耦合作用,本研究利用原子力显微镜(AFM)系统地测定了PEO电解质中富PEO相的机械性质。图4D显示,当富PEO相的[LiTFSI]分别降至0.80和0.44 M时,富PEO相的模量迅速上升至1.2 ~ 1.8 GPa。进一步研究I+富PEO两相区的富PEO相杨氏模量(图3A)可以看出,当[LiTFSI]小于0.8 M时,模量通常在1 GPa以上。由于纯PEO的杨氏模量为5-7 GPa,因此,在有限盐/增塑剂的半晶化富PEO层中,其杨氏模量可以达到1-2 GPa。上述结果表明,浓差极化诱导的锂沉积稳定机制是有效的。
相场模拟结果也证实了上述抑制机制的合理性,该模拟考虑了机械力-化学耦合。基于上述结果,模拟假设在[LiTFSI]《0.85 M时形成富PEO相,富PEO相的杨氏模量为1.6 GPa,而[LiTFSI]0.85 M区域的模量为1 MPa。由于在HCPE中没有形成新相,锂晶须在软的各向同性体聚合物电解质中快速生长(图4E)。相反,当[Li+]极化至0.85 M以下时,会形成富PEO相,有效抑制锂晶须的生长(图4F)。沉积的锂均匀,表面[Li+]不均匀性较低。这些模拟结果与实验结果非常吻合,证明高杨氏模量富PEO相的形成能够抑制晶须生长。
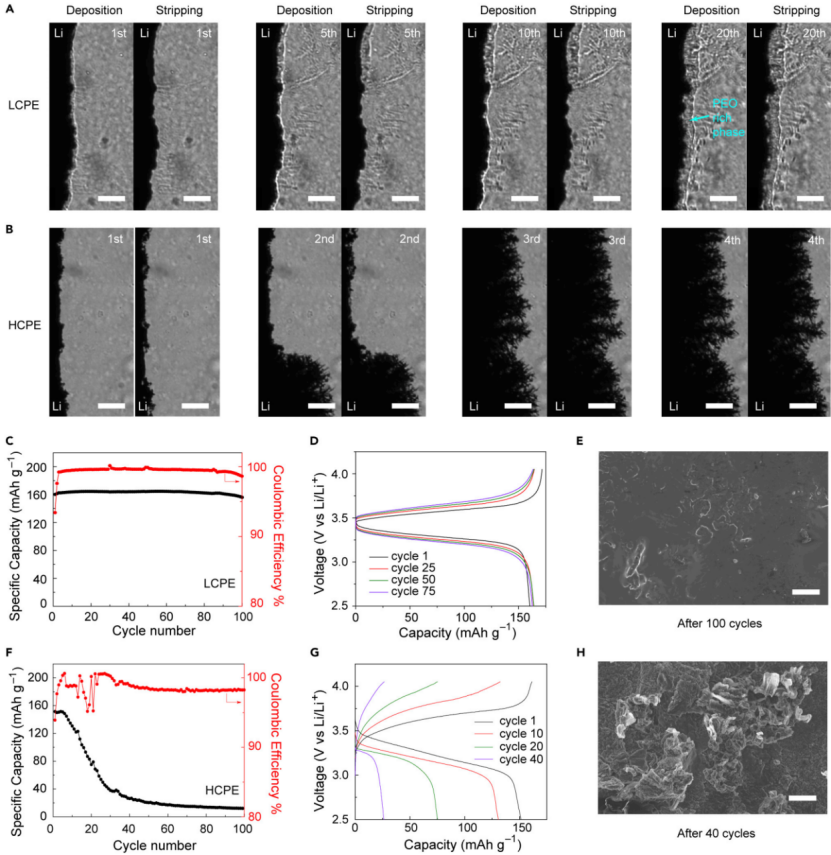
【图5】0.5 mA cm-2下,在(A)LCPE和(B)HCPE中锂电极电镀和剥离的明场图像。比例尺为100 μm。具有LCPE的LiFePEO4/Li电池(C)循环性能和(D)相应的充放电曲线。(E)循环100次后金属锂表面的SEM图像。具有HCPE的LiFePO4/Li金属电池(F)循环性能和(G)相应的充放电曲线。(H)循环40次后金属锂表面的SEM图像。(E)和(H)中的比例尺为10 μm。两个电池的电流密度为0.15 mA cm-2。所有电池在40℃下测试。
接下来,研究了这种策略在Li/Li电池和全电池循环中的有效性。首先,使Li/SN-LCPE/Li对称电池在0.5mA cm-2@0.25 mAh cm-2下循环20次。第一次沉积时没有形成明显的锂晶须,锂突起被不断增长的富PEO相冻结,锂沉积稳定。在锂剥离过程中,富PEO相和金属锂电极均收缩,不形成任何死锂。20次循环后,锂表面略有前移,表明这种抑制机制在多次循环后都是有效的。相比之下,如果没有富PEO相,使用HCPE的Li/Li电池在前几个循环内可以观察到快速的晶须生长,在重复剥离过程中形成大量的死锂(图5B)。
图5C显示,使用LCPE组装的LiFePO4/PEO/Li全电池实现了稳定的循环。初始放电容量为160.2 mAh g-1,CE为93.4%,在第10个循环中由于活化,容量缓慢增加到164.2 mAh g-1。100次循环后,容量为156.1 mAh g-1,容量保持率为97.4%。第5个循环到第100个循环的平均CE为99.5%(图5C)。充放电曲线显示,内阻只略有增加,没有出现晶须引起的短路现象(图5D)。SEM进一步显示,100次循环后,金属锂表面相对平坦,偶尔出现岛状形貌,表明富PEO相抑制锂晶须的有效性(图5E)。
另一方面,具有HCPE的LFP/PEO/Li电池很快失效,40次循环后容量从151.5 mAh g-1下降到26.2 mAh g-1(图5F)。充放电曲线显示,过电位急剧增加,表明可能存在晶须生长(图5G)。SEM图(图5H)证实,CE平均值仅为98.2%,这可能是由于锂晶须生长旺盛所致。
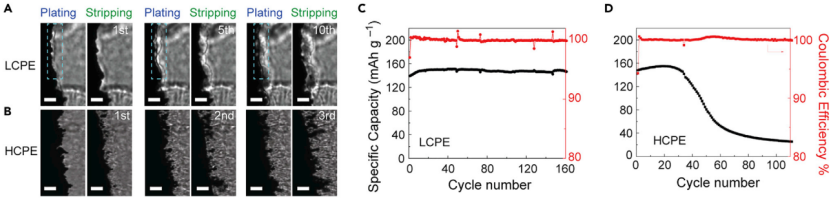
【图6】采用PC/LiDFOB-LiBF4双盐基LCPE和HCPE制备的Li/Li电池和LFP/Li电池中金属锂负极的循环稳定性(A和B)采用(A)LCPE和(B)HCPE制备的Li/Li电池在0.75 mA cm-2@0.375 mAh cm-2下循环时,锂负极的明场像。具有(C和D)LCPE和(D)HCPE的LiFePO4/Li金属电池在38℃下的循环性能。电流密度为0.3 mA cm-2。在LCPE中,PEOLiBF4=121, PC为PEO的90 wt%。在HCPE中,PEOLiBF4=61, PC是PEO的90 wt%。
这种相分离诱导晶须抑制在PEO电解质中是普遍存在的。在另一种二氟硼酸锂(LiDFOB)和四氟硼酸锂(LiBF4)溶解在PEO/碳酸丙烯酯(PC)增塑剂的体系中,也观察到类似的现象。首先确定了该体系中单相区和两相区的边界,即EO:Li+=6处。因此,在0.75 mA cm-2@0.375 mAh cm-2下对Li/PC-LCPE/Li电池进行测试,并使用光学显微镜对其进行表征。在锂沉积过程中观察到富PEO层,该层能够抑制锂晶须(图6A)。相比之下,EO/Li+=3的PC-HCPE没有出现相分离,形成了明显的晶须和死锂(图6B)。图6C显示,LFP/PC-LCPE/Li电池在0.3 mA cm-2能够稳定循环超过160次。而用PC-HCPE替代PC-LCPE时,100次循环后电池容量保持率仅为17.3%(图6D)。
4、总结与展望
本工作利用具有高时间和空间分辨率的SRS显微镜,首次观察到聚合物电解质中的动态浓差极化、相变以及它们与锂沉积的相关性。结果表明,浓差极化会诱导聚合物电解质中的相变,以及电极/电解质界面上富PEO相的形成。这种相变也存在于其他各种聚合物电解质体系中。这种新相的杨氏模量高达3 GPa,通过在锂负极上充当可逆的、自增强的保护层,有效地抑制了锂晶须的生长。相比之下,没有这种相变,传统的聚合物电解质具有较小的模量《1 MPa,锂晶须快速生长。基于上述结果,本工作提出了聚合物电解质的设计原则:在PEO-盐-增塑剂相图中,电解质的组成应该在单相和两相区域的边界处,这样电流就可以很容易地降低金属锂表面的盐浓度,从而在金属锂表面形成富PEO相。利用该机制,LFP/PEO/Li电池能够稳定循环,而没有该机制的电池在10个循环内迅速失效。该策略也与目前的电池材料制造工艺相兼容。此外,该策略对不同的盐和增塑剂具有普适性和有效性,有利于开发具有高热稳定性和能量密度的固体聚合物电解质基LMBs。
审核编辑 :李倩
-
电解质
+关注
关注
6文章
821浏览量
20203 -
固态电池
+关注
关注
10文章
709浏览量
27988
原文标题:哥伦比亚大学杨远Joule:反其道而行之,浓差极化对固态电池竟是好事?
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
清华大学:自由空间对硫化物固态电解质表面及内部裂纹处锂沉积行为的影响
研究论文::乙烯碳酸酯助力聚合物电解质升级,提升高电压锂金属电池性能

清华深研院刘思捷/港科大Kristiaan Neyts最新AEM封面文章:硫化物复合固态电解质

陈军院士团队最新Angew,聚合物电解质新突破

半互穿网络电解质用于高电压锂金属电池
北京科技大学范丽珍教授团队In和F共掺杂LPSCl制备固体电解质

一种创新的超薄固体聚合物电解质
固态电池中复合锂阳极上固体电解质界面的调控






 工商网监
工商网监
评论