 详细研究LIB和SIB的可逆相变
详细研究LIB和SIB的可逆相变
背景介绍
在锂离子电池和钠离子电池(LIBs和SIBs)中,氧化物层状正极的氧氧化还原(OR)通常被用于提高能量密度。
起初,研究人员首次在一种典型的锂过量锰基层状氧化物(Li2MnO3)中发现并利用了这种OR反应,在约4.5 V时,其容量超过300 mAh/g。在基本反应框架中,氧化还原失活的Mn4+离子在充电时触发氧的氧化,其电子结构来源于[Li1/3Mn2/3]O2层中过量的Li离子。虽然具有良好的阴离子特性,但在随后的放电过程中,氧的电荷曲线并没有被保留,同时伴随着电压下降和容量下降。
尽管通过掺杂、涂层和构建复合材料,能够一定程度上改善OR可逆性,但仍然存在严重的挑战,因为研究人员尚不知道如何可持续地利用Li2MnO3的纯阴离子氧化还原容量。因此,需要新的方法来释放Li2MnO3的OR活性。
正文部分
1、成果简介
近日,韩国庆熙大学Duho Kim团队,提出了一种交互设计概念,模拟SIB中锂过量Na氧化物表现出的非滞后可逆(nHR)阴离子活性,以充分利用LIB中Li2MnO3的氧来提供容量。当Li离子位于Na层状氧化物荷电结构的四面体位置时,在钠化/脱钠过程中,层间滑移和氧氧化还原会引起可逆的堆叠序列转变。有趣的是,F掺杂Li2MnO3的堆叠序列转变也是可逆的,脱锂相不存在O−O二聚体,其Li构型与上述Na正极一致。虽然Mn的面内迁移导致分子O2被捕获在体相中,但在正常的脱锂模式下,电子结构表现出可逆的晶格O(2p)活性。
2、研究亮点
本工作利用第一性原理计算,详细研究了LIB和SIB的可逆相变。结果发现,它们形成的相都没有O−O二聚体出现,因为Li和Na层的四面体位置都被Li占据。正常脱锂模式下阳离子和阴离子的电子结构表现出可逆的O2−/On−氧化还原活性,而Mn的面内迁移导致了Mn−2O2−Mn基团的形成。
3、图文导读
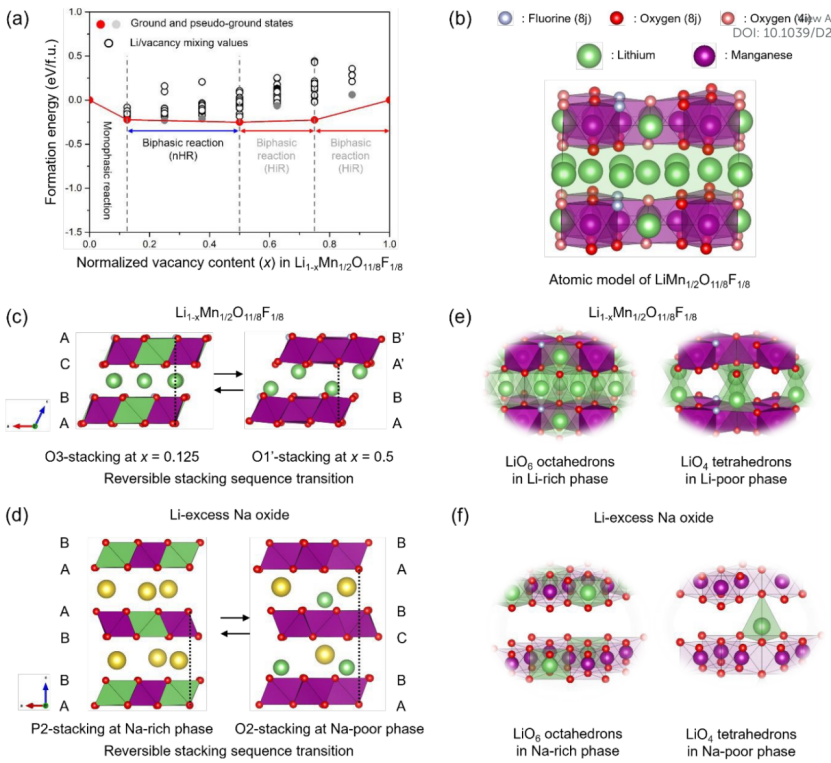
【图1】(a)混合焓的形成能,考虑所有可能的Li离子及其空位与整个Li1−xMn1/2O11/8F1/8中空位含量(x)的比值(0.0≤x≤1.0)。(b)包含Li、Mn、O和F离子的原子模型。阴离子位点被分为8j和4i位点。(c)在[010]投影图中,x=0.125和0.5时的Li1−xMn1/2O11/8F1/8原子结构。(d)P2型锂过量Na氧化物正极中富Na相和贫Na相的[010]投影图。(e-f)富A相和贫A相中详细的Li构型,其中A分别指Li和Na。
图1a显示了混合焓的形成能,考虑所有可能的Li离子及其空位与整个Li1−xMn1/2O11/8F1/8中空位含量(x)的比值(0.0≤x≤1.0),其完全锂化结构如图1b所示。由于Li2MnO3中的氧氧化被认为能够补偿Li脱出引起的电荷不平衡,因此推测阴离子位的F掺杂部分降低了Mn的价态(从Mn4+到Mn3+)。这将在宿主材料的初始脱锂过程中触发Mn3+/Mn4+的阳离子氧化还原反应。
因此,接下来按照图1a中红线所示,研究整个脱锂过程中的热力学相稳定性。能量途径表明,Li1−xMn1/2O11/8F1/8在0.0≤x≤0.125范围内发生单相反应,这可能是由Mn氧化还原反应诱导的。从热力学的角度来看,在掺F氧化物中的单相反应与在Li2MnO3中是不同的,因为在充电时氧氧化引起了一个严重的双相反应。
进一步脱锂到x=0.5产生了第一个双相反应,双相由Li0.875Mn1/2O11/8F1/8和Li0.5Mn1/2O11/8F1/8组成。上述结果表明,在0.125≤x≤0.5时的双相反应可以反映出充电(放电)时的nHR氧容量,因为掺F氧化物的热力学行为与LiFePO4相似,表现为可逆的两相反应。
更重要的是,空位含量中的氧活性在补偿锂脱出引起的电荷不平衡方面发挥了重要作用。Li1−xMn1/2O11/8F1/8的第一个双相反应可以用组合相(CP)混合焓来解释。当0.5≤x≤0.75时发生了第二次双相反应,当x>0.75时,脱锂导致第三个双相反应。这些双相反应是由x=0.625和0.875时存在的伪基态决定的。
不幸的是,这些相是不稳定的,因为参考线上的相混合焓形成能比第一次双相反应中的相要高得多,因此导致了迟滞和不可逆(HiR)的氧容量。也就是说,与0.125≤x≤0.5相比,第二和第三区域的CP混合焓表现出非常不稳定的值。这导致相分离速度快得多。
基于对热力学相稳定性的深入研究,很容易得出结论:Li1−xMn1/2O11/8F1/8在整个空位范围内发生了多次相变。而未掺F的材料在充电时迅速发生严重的相变。
考虑到钠基层状氧化物在脱钠过程中通常会发生多个相变,其反应被认为是堆叠类型的变化(即Pn−On相变,其中n可以变化),因此研究了Li1−xMn1/2O11/8F1/8在x=0.125和0.5处的详细原子结构,以确定双相反应中涉及的堆叠类型转变。图1c显示了[010]投影图中相应的原子结构。起点相为O3型层状结构,而终点的堆叠类型变为O1’型结构。
也就是说,通过层间滑移诱导的O3−O1'可逆堆叠序列转变可能是一种热力学反应机制,类似于锂过量Na氧化物中氧氧化时可逆层间滑移导致的P2−O2堆叠序列转变。图1d显示,富钠相的堆叠类型为P2型层状结构,而贫钠相的为O2型结构。虽然在初始点的Li和Na氧化物表现出不同的结构类型,但在终点的Li和Na氧化物表现出类似的层间滑移诱导的堆叠序列转变。
因此,借鉴Li过量Na氧化物中层间滑移诱导堆叠序列转变的概念,有望通过F取代在第一个双相反应中激活Li2MnO3中的全部氧活性。为了确定Li1−xMn1/2O11/8F1/8中Mn在0.125≤x≤0.5时不向LiO2层迁移的前提,本工作研究了不同相的锂离子排序。图1e显示了[100]投影视图中富锂(x=0.125)和贫锂(x=0.5)相的原子结构。所有锂离子都占据了富锂相LiO2和Mn层的八面体位置。而在贫锂相中观察到不同的Li构型,其在整个晶体框架上看起来很有序。也就是说,贫锂相中的所有锂离子都明显位于四面体位置,而锰层中没有锂离子。
因此,可以推断出Li2MnO3层间的Mn迁移机制,即Mn离子通过四面体位置迁移到八面体位置。沿着这条线,四面体位置上的有序Li离子可能在第一次双相反应中对抑制Mn离子迁移起了决定性作用。为了进行比较,图1f显示了锂过量钠氧化物中富钠相和贫钠相的锂离子排序。同样地,在O2型贫钠相中发现了锂离子在四面体位置的异常排列,并且阳离子不倾向于占据Mn层中的八面体位置。而P2型富钠相中锂离子倾向于占据八面体位置,这与掺F锰氧化物贫锂相中的锂位置相似。
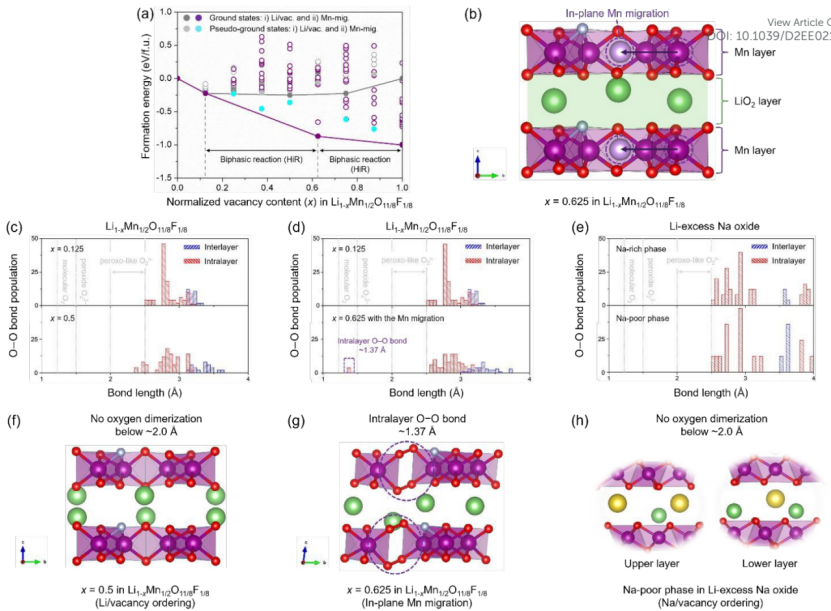
【图2】(a)在Li/空位和面内Mn(ip-Mn)迁移模式下混合焓的形成能,考虑所有可能的Li离子及其空位与整个Li1−xMn1/2O11/8F1/8中空位含量(x)的比值(0.0≤x≤1.0)。(b)面内Mn迁移示意图。(c)根据x=0.125和0.5处的Li/空位排序,(d)掺F氧化物中x=0.125和0.625处的ip-Mn迁移,以及(e)锂过量Na氧化物中富Na相和贫Na相的Na/空位排序,将O−O数分为两部分,i)层间和ii)层内。(f-h)每个带电结构中沿a轴的相应原子结构。
除了Mn层间迁移外,阻碍OR的关键因素之一是Li和Na层状氧化物中Mn层内的面内Mn(ip-Mn)迁移。然而,这一问题在掺氟模型中没有得到解决。因此,接下来计算了ip-Mn迁移下Li1−xMn1/2O11/8F1/8中所有可能的Li离子及其空位的形成能(0.0≤x≤1.0),如图2a所示。计算模式如图2b所示。能量图显示,当x<0.375时,Li/空位模式的相稳定性高于ip-Mn模式,而当x>0.375时则低于ip-Mn。在x<0.125范围内发生了单相反应,但在0.125≤x≤0.625范围内发生了双相反应,双相由Li0.875Mn1/2O11/8F1/8和Li0.375Mn1/2O11/8F1/8组成。
与Li/空位模式相比,该反应是由在x=0.625处存在的稳定相决定的。此外,ip-Mn模式下,在x=0.25, 0.375和0.5处的伪基态比Li/空位模式下第一个双相反应的伪基态更不稳定。此外,在ip-Mn模式下,进一步的脱锂(x>0.625)导致在Li1-xMn1/2O11/8F1/8中发生第二次双相反应,其热力学相稳定性与第一个双相区相似。
为了了解ip-Mn模式下的严重双相反应,研究了Li1−xMn1/2O11/8F1/8在x=0.125和0.625处的详细原子和电子结构。由于O-O二聚体的形成是在OR基氧化物正极中诱导HiR氧容量的关键因素,因此根据Li/空位排序和ip-Mn迁移模式,计算了每种模式下第一双相区域的O-O键数量。图2c显示,当x=0.125时,层内的O−O基团键长最短(~2.53 Å),在双相反应过程中减少到~2.38 Å。
也就是说,在Li/空位模式下,层间和层内均未在2.0 Å以下形成O−O键,表明过氧化物O22−(1.48 Å)或分子O2(1.21 Å)没有在晶体框架上形成。这些结果表明,Li1−xMn1/2O11/8F1/8在0.125≤x≤0.5条件下发生了基于晶格O2−/On−(n<2)的理想氧氧化过程。
另一方面,ip-Mn模式下,在脱锂结构(x=0.625)层内,发现了最短的O−O基团(~1.37 Å),如图2d所示。这意味着在Mn层中产生一个空位使带电结构不稳定,导致ip-Mn迁移到最近的空位位置。因此,Mn的迁移导致Mn层内空位团簇的形成,从而使分子O2被捕获在体相中,来实现稳定的氧氧化。
类似地,充电时,锂过量钠氧化物正极中也观察到理想的氧氧化行为,如图2e所示。每种模式下,x=0.5和0.625时Li1−xMn1/2O11/8F1/8以及Li过量Na氧化物中贫Na相的原子结构如图2f-h所示。
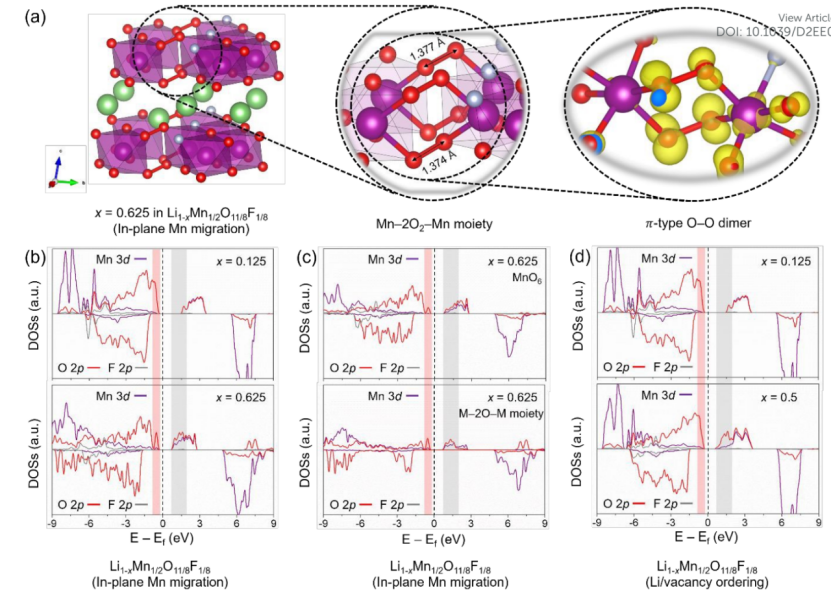
【图3】(a)Li1−xMn1/2O11/8F1/8在x=0.625处面内Mn迁移模式下的详细原子结构,以及价带边缘Mn−2O2−Mn构型和相应电子密度的放大图像。(b)面内Mn迁移模式下x=0.125和0.625处Mn(3d)、O(2p)和F(2p)电子的部分态密度(PDOSs)分布图,(c)将图3b中x=0.625时Li1−xMn1/2O11/8F1/8的PDOSs分解为MnO6和M−2O−M两个部分。(d)在Li/空位排序中,x=0.125和0.5处的PDOS。
图3a显示了ip-Mn条件下Li1−xMn1/2O11/8F1/8在x=0.625处的详细原子结构,其放大图像显示存在被体相捕获的O2分子。对应的O2二聚体(~1.37 Å)与图2e中最短的O−O基团一致,在带电正极中产生了Mn−2O2−Mn(M−2O−M)构型。为了从根本上理解这种含O2构型,计算了Mn(3d),O(2p)和F(2p)离子在充电时的电子结构,其取决于脱锂模式。
图3b为ip-Mn条件下Li1−xMn1/2O11/8F1/8中阳离子和阴离子在x=0.125和0.625处的部分态密度(PDOSs)分布。Mn离子在起始点的PDOS是典型的Mn4+价态,它的电子构型在充电时触发了氧氧化。由x=0.625时的PDOS可以看出,进一步的脱锂并没有改变Mn4+的荷电状态,而氧化的O(2p)电子出现在导带中。这些结果表明,氧离子是x>0.125时电荷补偿机制的关键因素。
为了深入了解M-2O-M构型,将x=0.625处的PDOS分解为两个组分:i)MnO6和ii)M-2O-M,如图3c所示。与MnO6相比,M−2O−M构型的PDOS有明显的差异:红色区域的O(2p)强度发生变化,灰色区域氧化O(2p)电子数增加。前者与体相捕获的O2分子有关,可直接归因于π型O−O二聚体的空间电子密度。
此外,在主体材料进一步充电时,几乎没有任何可氧化的氧。也就是说,O2分子的形成不仅减少了能氧化还原的O(2p)离子,而且可能加速了x>0.625时Li1−xMn1/2O11/8F1/8的结构破坏。为了进行比较,对Li/空位模式下x=0.125和x=0.5处的PDOS进行了检查,如图3d所示。
在后一种带电结构中,在费米能级以下的O(2p)电子数总体上与前一种结构相似,但在灰色区域可以发现氧化O(2p)电子数发生变化。有趣的是,这与ip-Mn模式下的氧化行为一致,但在x=0.5时,没有观察到尖锐的O(2p)峰,其代表含O2基团的形成。由于缺少可逆的氧电子,该基团与不可逆氧容量高度相关。
4、总结和展望
为了充分利用Li2MnO3脱锂过程中的纯阴离子容量,本工作提出了一种交互式设计概念,模拟锂过量钠氧化物中非滞后和可逆的氧氧化还原特性。另外,利用第一性原理计算,构建了关键的设计框架,从而能够可持续地利用氧提供容量,如多相转变、由层间滑移引起的可逆堆叠序列转变,以及在四面体位置上迁移锂离子。
从局部结构的角度来看,由于Li离子占据四面体位置,充电状态下的F掺杂Li2MnO3和锂过量Na氧化物没有观察到O−O二聚体的形成。Li/空位脱锂模式下的电子结构表现为基于晶格O2−的可逆氧氧化,而面内Mn迁移模式下的电子结构表现为O2-捕获。这种在LIB和SIB之间通用的交互设计理念有望实现非迟滞和可逆的氧氧化还原,从而极大提高电池能量密度。
审核编辑:刘清
-
锂离子电池
+关注
关注
85文章
3242浏览量
77763 -
钠离子电池
+关注
关注
6文章
219浏览量
14732
原文标题:EES:如何可持续利用氧提供电池容量?
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
课题组JACS:O3-NaNi1/3Fe1/3Mn1/3O2的结构演变解析

相变材料在电池散热系统中的应用研究

DSC测试相变储能材料相变温度
差示扫描量热仪DSC测试相变储能材料相变温度

请问单相变频器输出到uvw怎么接线?
单相转三相变频器的缺点
三相变频器接220v能不能调试
三相变频器改220v单相方法怎么接线
三相变频器正反转怎么接线
深入解析相变器件原理及超薄均热板电池应用
AG32在VSCode下使用lib库
单相变压器的结构组成与工作原理

什么是相变存储器?如何表征相变材料及器件电学性能?





 工商网监
工商网监
评论