 亚稳1T-MoS2中的电荷自调节效应可以调控电子活性态
亚稳1T-MoS2中的电荷自调节效应可以调控电子活性态
【研究背景】
六方结构二硫化钼(2H-MoS2)因其成本低、活性适中而被认为是一种很有前途的贵金属析氢反应催化剂的替代品。理论计算和实验观察表明,2H-MoS2的HER活性主要归因于费米能级附近的电子态优化了氢吸附强度。然而,传统的2H-MoS2缺乏充足的电子活性态。到目前为止,已经进行了多种优化策略来调节电子状态,如异构原子掺杂、缺陷工程、相变和畴边界。
然而,MoS2电催化性能仍然不能令人满意。体相2H-MoS2本质上是一种间接隙半导体(带隙Eg = 1.29 eV;室温下的电子电导率σRT=~10−4 S cm−1)。相比之下,亚稳三角形1T'''-MoS2是一个窄隙半导体(Eg=0.65eV,σRT=2.26 S cm−1)。1T'''-MoS2结构中丰富的Mo−Mo键可以作为电子储层,调节费米表面的电子态,控制氢的吸附/解吸。1T'''-MoS2有望成为一种很有前途的HER电催化剂。
【成果简介】
中国科学院上海硅酸盐研究所,上海材料基因组研究所 中国科学院大学,北京大学的黄富强和刘建军教授在酸性K2Cr2O7水溶液中通过拓扑化学反应和化学腐蚀合成了1T'''-MoS2大块晶体,其具有S空位的1T'''-MoS2 (1T'''-MoS2-Vs)。在10mA cm−2处的过电位为158mV, Tafel斜率为74.5mV dec−1,循环稳定性良好,远远优于无S空位的2H-MoS2。
【研究亮点】
1. S空位通过形成悬空键激活Mo−Mo键, 2. 激活的Mo−Mo键可以通过增强S和H原子之间的相互作用调节电子态,促进质子吸附。
【图文导读】
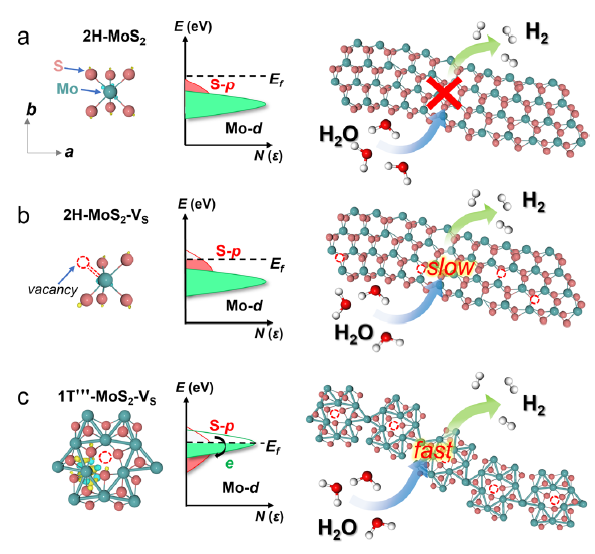
图 1 a 2H-MoS2,b不含Mo−Mo键的2H-MoS2-Vs和c 1T "'-MoS2-VS的电子态。
MoS2的基面活化如图1a-c所示,分别说明了2H-MoS2、2H-MoS2-Vs和1T'''-MoS2-VS的活性电子态的差异。由于费米表面是孤立的,活性电子态的缺乏导致2H-MoS2平面的惯性(图1)。2H-MoS2与2H-MoS2-Vs的轨道在相对较深的能级杂化,这不利于在费米表面附近提供额外的电子态供质子吸附(图1b)。
相反,在1T'''-MoS2-VS中,Mo−Mo的有源电子态穿过费米表面的键和S原子,导致从激活的Mo−Mo键到S原子的电荷转移(图1c)。活性Mo−Mo键可诱导电荷重新分配,调节S原子的电子态,从而增强S−H键的强度。
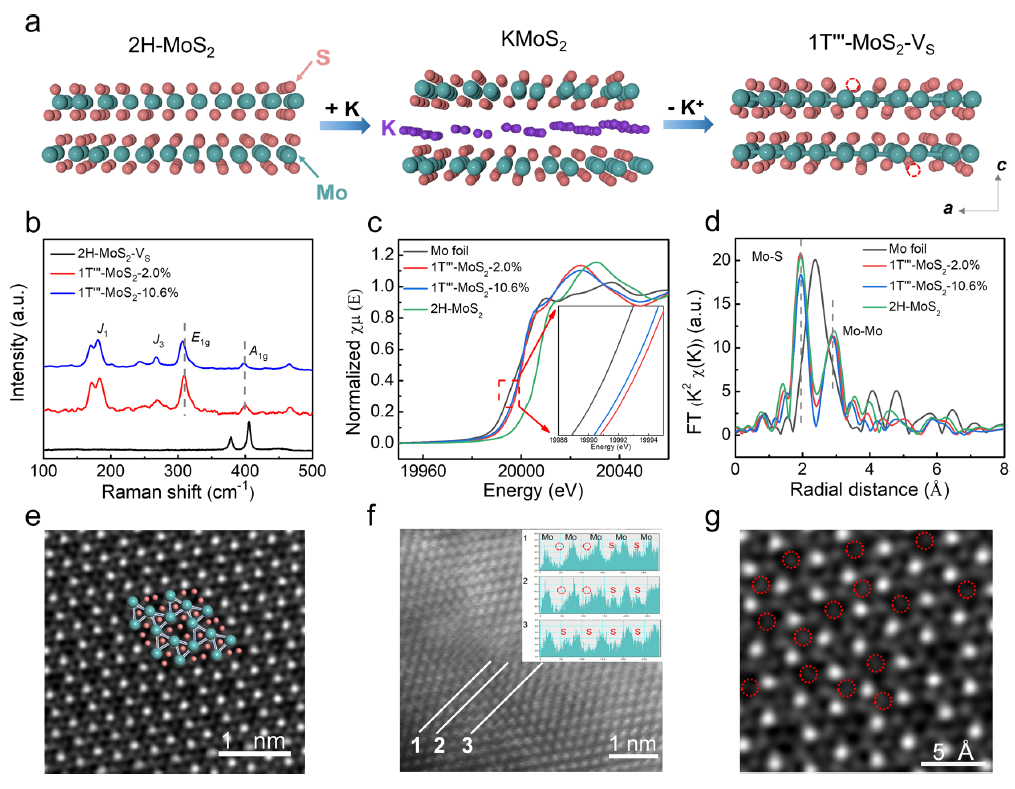
图2 a 高温固相反应合成的1T"'-MoS2-VS晶体的示意图 b 2H-MoS2-VS, 1T"'-MoS2−2.0%和1T"'-MoS2−10.6%的拉曼光谱。c 钼k边XANES光谱和d FT-EXAFS光谱。e 1T "'-MoS2−10.6%的HAADF-STEM。f 1T "'-MoS2−10.6%中S空位的ACTEM。
如图2a所示。高温固相反应合成的KMoS2晶体的中间层中含有高度有序的K+离子。在将Mo (III)氧化为Mo (IV)的K+离子萃取过程中,KMoS2中的Mo−Mo键部分保持为1T'-MoS2和1T'''-MoS2体块。在酸性K2Cr2O7溶液中进一步氧化1T'''-MoS2,通过不同的刻蚀时间,得到不同浓度的S空位。
采用1T'''-MoS2−10.6%的高温真空退火法制备了2H-MoS2-VS。从拉曼光谱 (图2b)可以看出,1T'''-MoS2−2.0%的6个特征峰分别位于177、243、267、305、398和463 cm−1。随着S空位的增加,1T'''-MoS2−10.6%的E1g峰移至302 cm−1。
1T'-MoS2的拉曼光谱有四个特征峰,2H-MoS2-VS的E1g峰与1T'''-MoS2-VS样品的特征峰完全不同,这是由于它们的晶体结构不同。如图2c表明1T'''-MoS2−2.0%和1T'''-MoS2−10.6%具有相似的吸收边和白线峰,但与2H -MoS2和Mo箔不同。图2d显示1.9和2.8 Å处的两个特征峰归属于Mo−S和Mo−Mo键。
1T'''-MoS2−10.6%在S空位浓度较高时,未观察到明显的位移,表明化学腐蚀前后Mo−Mo键长基本不变。2H-MoS2的特征峰(2.9 Å)可归因于较长的Mo−Mo距离。而1T'''-MoS2−10.6%的Mo−S键峰值强度降低,而Mo−Mo键峰值强度保持不变,表现为配位数降低,可能是由于S空位增加后产生的缺陷增多所致。
特征峰向较低能量移动0.45 eV,表明在1T'''-MoS2−10.6%中Mo价态较低。在图2e中,1T'''-MoS2−10.6%的晶格结构高度扭曲。如图2f所示,通过像差校正透射电镜(ACTEM)检测了1T'''-MoS2−10.6%中存在S空位。1T'''-MoS2−10.6%的HAADF-STEM图像(图2g)进一步证实了相应浓度S空位的成功形成。
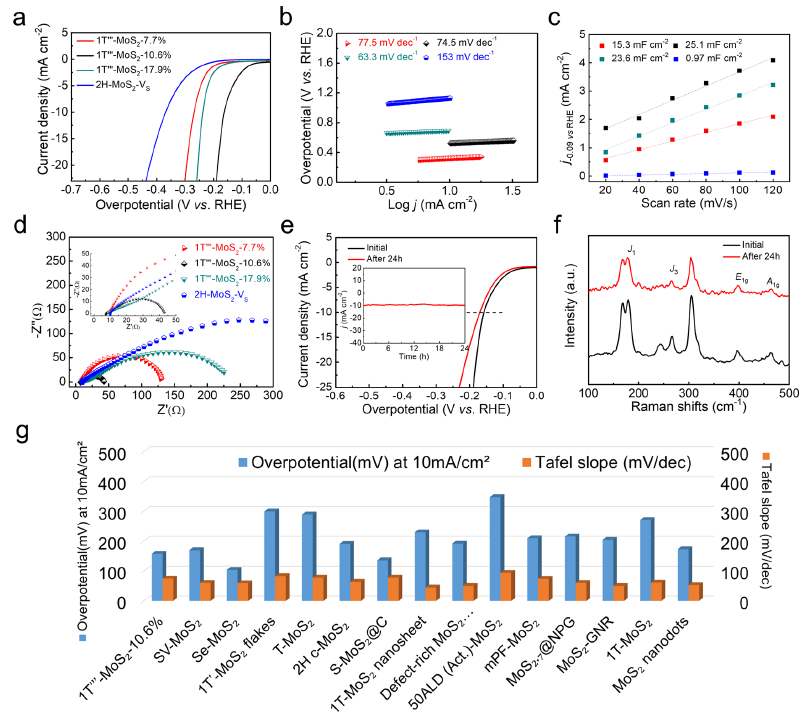
图3 a 样品的线性扫描极化曲线。b极化曲线对应的Tafel曲线。c电流密度与扫描速率的差值图,d Nyquist曲线。e1T"'-MoS2−10.6%耐久试验前后24h的LSV曲线。(f) 1T"'-MoS2−10.6%在24 h试验前后的拉曼光谱。g 1T"'-MoS2−10.6%和2H-MoS2-VS催化剂在10mAcm−2电流密度下的过电位。
如图3a所示。当氧化时间增加到1 h时,过电位下降到158mV (1T'''-MoS2−10.6%)。如图3b所示,1T'''-MoS2−10.6%的Tafel斜率为74.5mV dec−1,由图3c可知,1T'''-MoS2−10.6%的ECSA值最高(25.1 mF cm−2),显著大于对比样品,说明适当浓度的S空位可以显著扩大ECSA,暴露更多电化学活性位点。
如图3d所示,1T'''-MoS2−10.6%的样品Rct最低,说明适当的S空位可以加速HER电极动力学,减少欧姆损耗。1T'''-MoS21具有良好的稳定性 (图3e)。在电化学测试前后的拉曼光谱和XRD中没有观察到相变化 (图3f)。与最近报道的纳米结构MoS2电催化剂相比, 1T'''-MoS2−10.6%具有优异的电化学性能(图3g)。
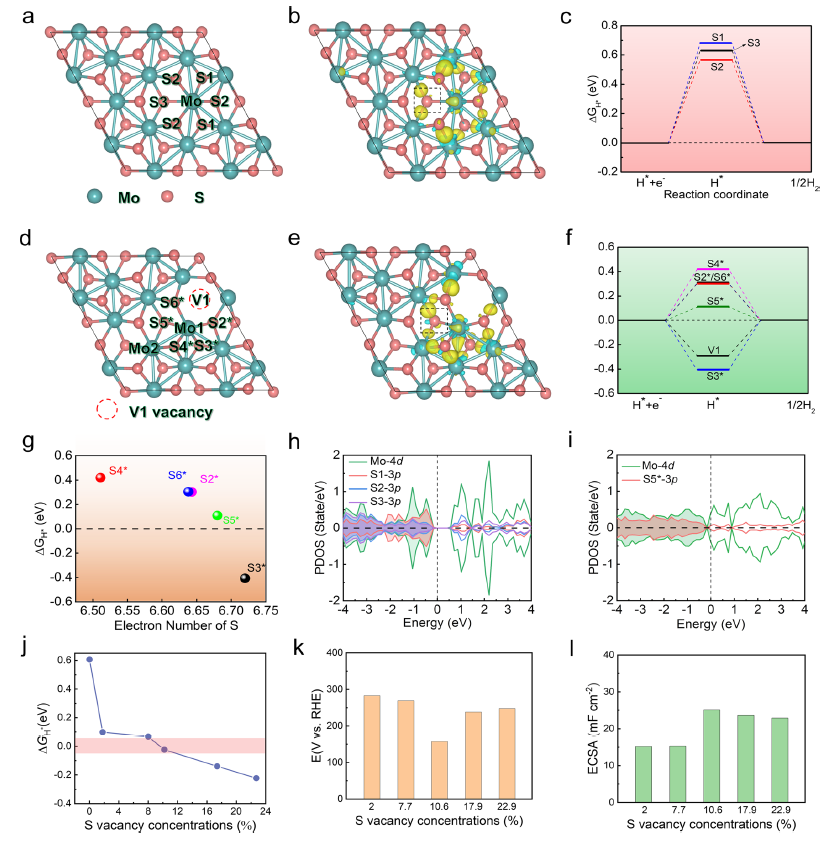
图4 a, d优化后的1T"'-MoS2, 1T"'-MoS2-V1的结构。b, e 1T"'-MoS2和1T"'-MoS2-V1中不同吸附S位点的电荷密度差。c, f1T"'- MoS2和1T"'- MoS2- V1中Mo周围不同暴露的S原子的ΔGH*。g ΔGH*与1T"'- MoS2-V1上S的电子数的相关性。h 1T"'- MoS2的S1、S2和S3原子的PDOS。i 1T"'- MoS2-V1 S5*原子的态密度。j S空位浓度与ΔGH*之间的相关性。k, l不同S空位浓度下1T"'- MoS2的过电位和ECSA。
图4a和b显示了1T'''-MoS2多个吸附位点的弛豫结构和电荷密度差。S和Mo原子之间存在几个固有的Mo−Mo键和扭曲的八面体配位(图4a)。计算得到的三个不同S吸附位点的电荷密度差如图4b所示,表明S原子的电子态较少。在三个不同的吸附位点上计算的ΔGH*值在0.567 ~ 0.682 eV之间(图4c),这不利于氢的吸附。如图4d-f所示,构建1T'''-MoS2模型中三个不同的空位和对应的电荷密度差,研究氢吸附行为与电子结构(1T'''-MoS2-Vn,n = 1-3)之间的关系。引入S空位后,通过形成悬空键来激活Mo−Mo键和邻近的S原子(图4 d)。
如图4 e所示,与原始的1T'''-MoS2相比,激活的Mo−Mo键的电荷调节作用使S原子的电子态增加,与激活的Mo−Mo键相连的S原子具有较强的键合强度 (图4f)。因此,1T'''-MoS2-V1中Mo−Mo键激活所引起的电荷再分配,导致V1空位周围S原子额外的电子,促进S−H键的形成。
如图4h所示,1T'''-MoS2由于带隙较大,电导率相对较差,含有激活Mo−Mo键的1T'''-MoS2-V1受电荷自调节作用,使费米表面带隙缩小,新隙态数量增加,这有利于电导率的提高(图4i)。最佳ΔGH*(−0.024 eV)与1T'''-MoS2-10.6%(图4j- l)的最佳性能一致。
【总结与展望】
综上所述,作者从理论上和实验上揭示了亚稳1T'''-MoS2中的电荷自调节效应可以调控电子活性态,进而优化HER活性。随着S空位的增加,激活的Mo−Mo键会进一步重新分配相邻S原子的活性电子态,从而促进氢吸附。电化学结果表明,最佳的1T'''-MoS2−10.6%在10mA cm−2电流密度下表现出158mV的过电位和74.5mV dec−1的Tafel斜率,远远优于2H-MoS2-VS,验证了激活Mo−Mo键对提高HER活性的电荷自调节作用。
本研究为开发具有良好能量转换和存储性能的氧化物/硫化物基催化剂提供了设计理念和策略。
审核编辑:刘清
-
XRD
+关注
关注
0文章
132浏览量
9074 -
拉曼光谱
+关注
关注
0文章
83浏览量
2738
原文标题:黄富强/刘建军Nature子刊: 富S空位的1T'''-MoS2电荷自我调节增强HER活性
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
MOS管在电池管理系统(BMS)中应用的解析

MOS管在开关电源中的应用及作用
MOS管击穿原理分析、原因及解决方法
电流流的是正电荷还是负电荷
P沟道MOS管的工作原理和导通条件
场效应管怎样调节电流
MOS管输入电阻很高,为什么一遇到静电就不行了?
了解压电传感器:压电效应
未调节的60 mA电荷泵电压逆变器TPS6040x-Q1数据表

基于JEDEC栅电荷测试方法测量MOSFET的栅电荷






 工商网监
工商网监
评论