 使用AlphaFold2进行蛋白质结构预测
使用AlphaFold2进行蛋白质结构预测
前言
AlphaFold 2,是DeepMind公司的一个人工智能程序。2020年11月30日,该人工智能程序在蛋白质结构预测大赛CASP 14中,对大部分蛋白质结构的预测与真实结构只差一个原子的宽度,达到了人类利用冷冻电子显微镜等复杂仪器观察预测的水平,这是蛋白质结构预测史无前例的巨大进步。这一重大成果虽然没有引起媒体和广大民众的关注,但生物领域的科学家反应强烈。
目前,AlphaFold2的源代码已经在GitHub上公开,而且现在科学家正在利用AlphaFold2对已有的蛋白数据库进行高通量的预测,建立了一些模式生物物种所有蛋白的AlphaFold2预测结构数据库。
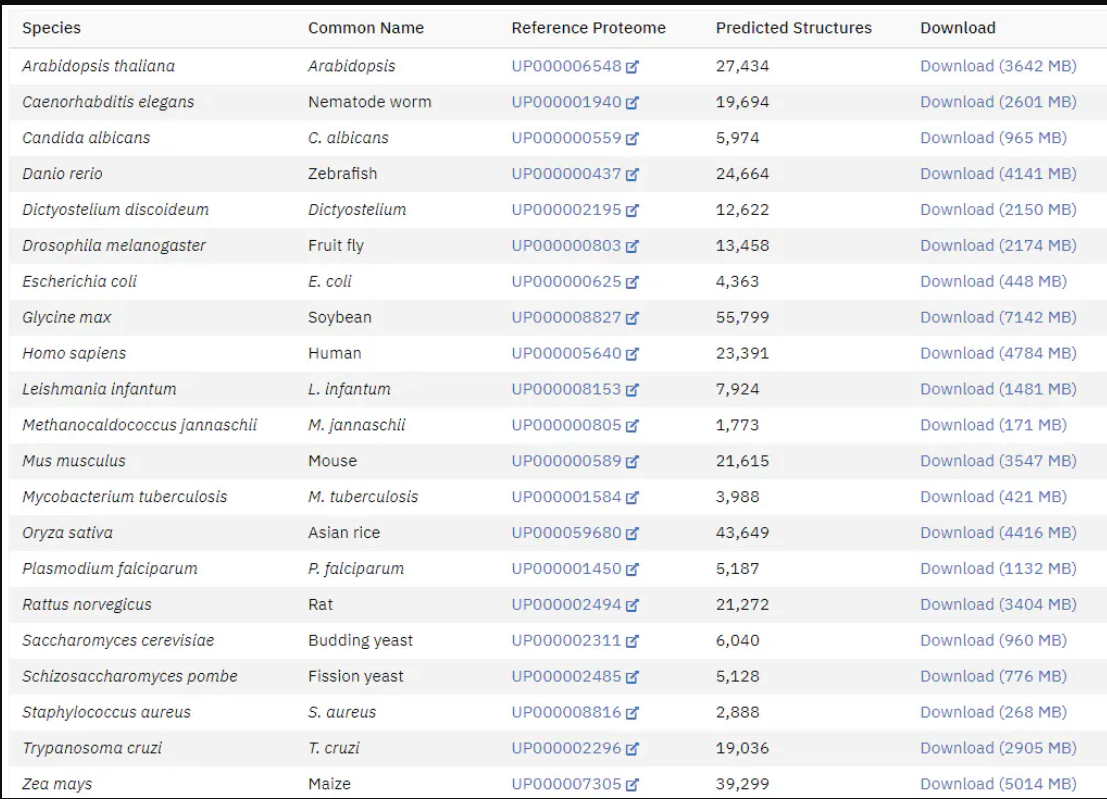
可以看到,虽然利用AlphaFold2预测了这么多生物的数据库,但是并未覆盖所有的蛋白序列数据库,所以只有搭建本地的AlphaFold2服务,你才能用AlphaFold2随心所欲的预测自己研究蛋白的结构。
接下来将给大家介绍AlphaFold2的使用方法,在北鲲云上免安装使用。对于没有Linux基础或本地硬件配置不足的人,仅需1分钟即可成功提交蛋白质结构预测任务,能够省去很多麻烦。
二、在北鲲云使用AlphaFold2进行蛋白质结构预测
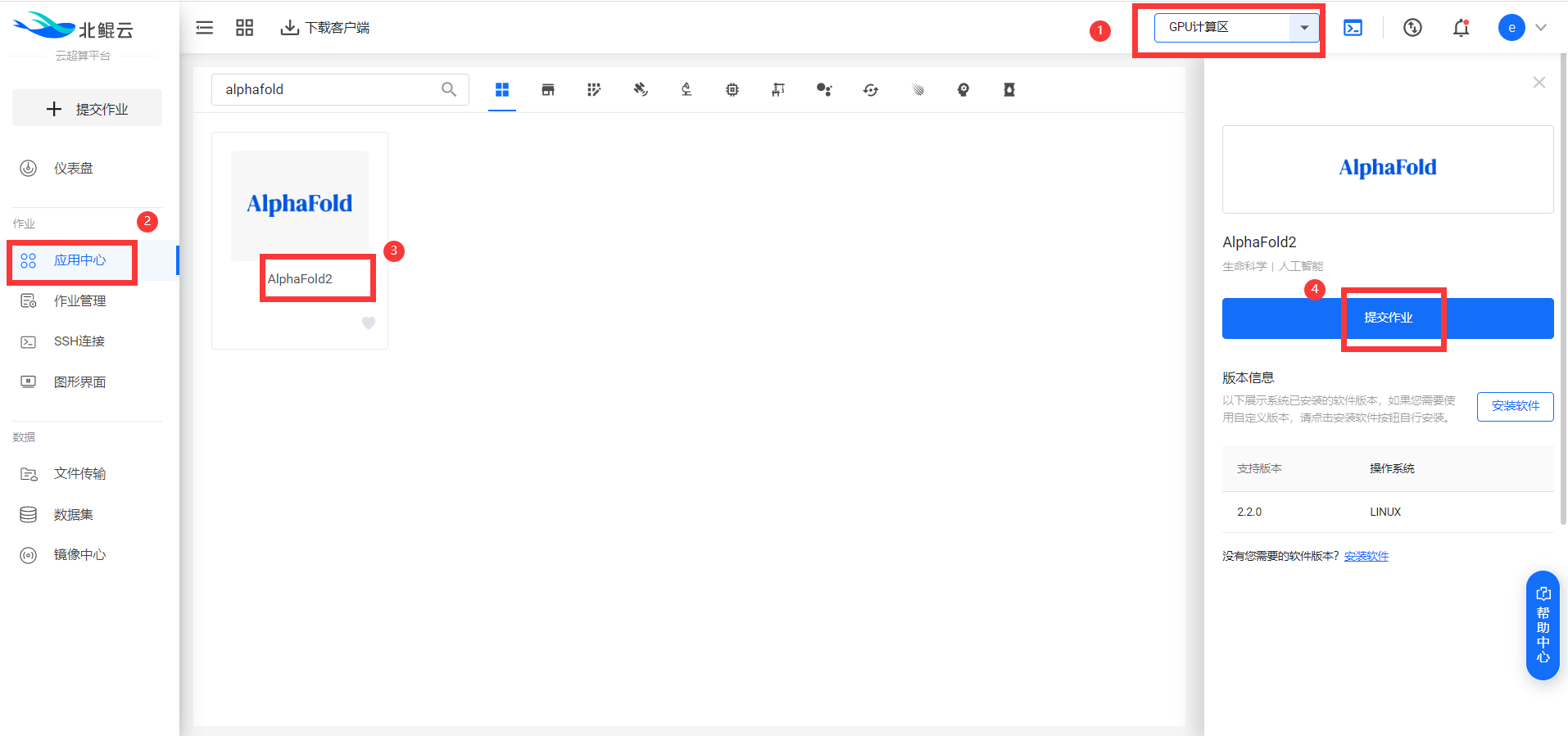
1. 选择AlphaFold2
在“应用中心”搜索AlphaFold2软件并选中,在右侧弹出的软件详情栏中点击“提交作业”。
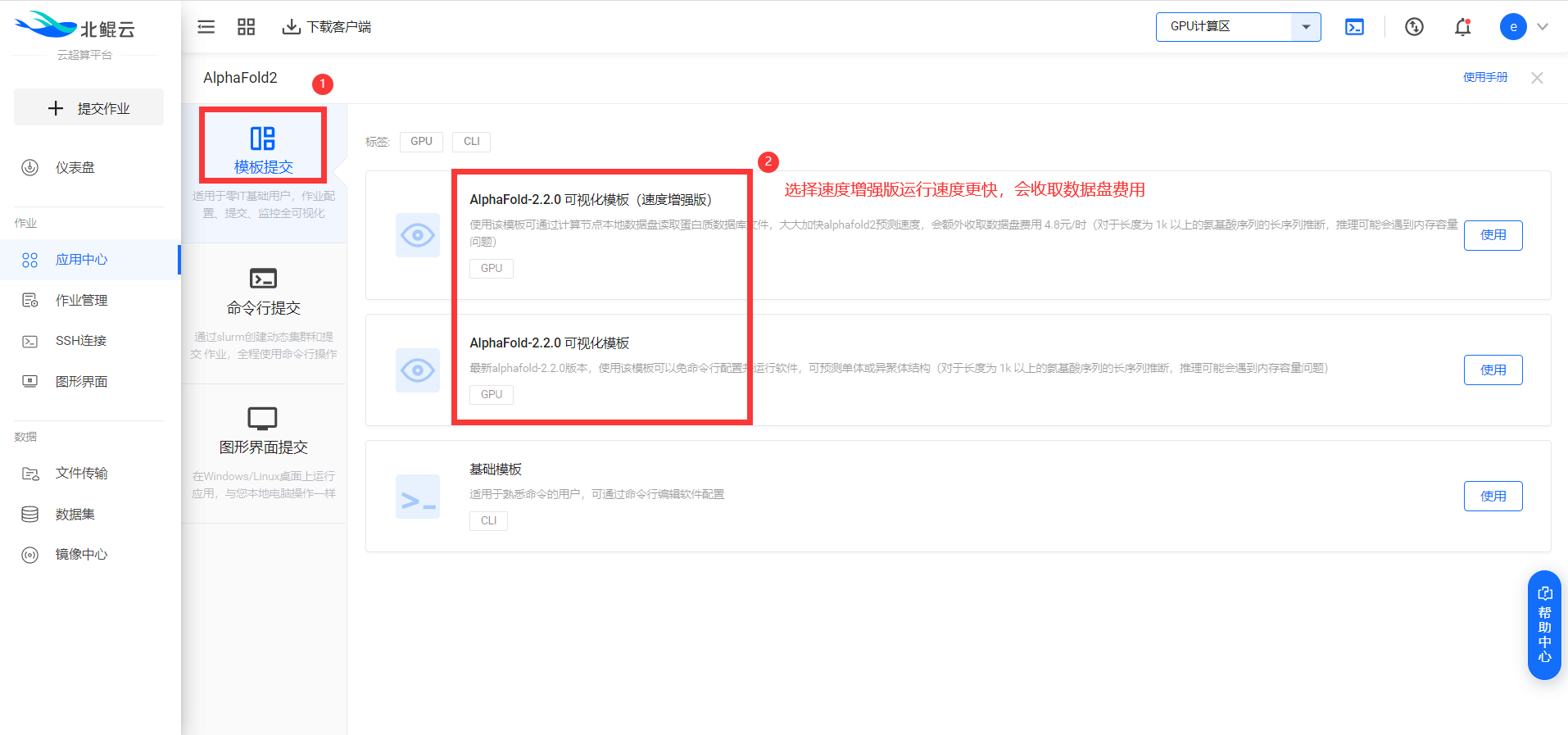
2. 选择可视化模板提交
推荐选择可视化“模板提交”的方式提交作业,平台已为AlphaFold2内置了几个可视化模板,按要求填写相应参数即可提交预测任务。
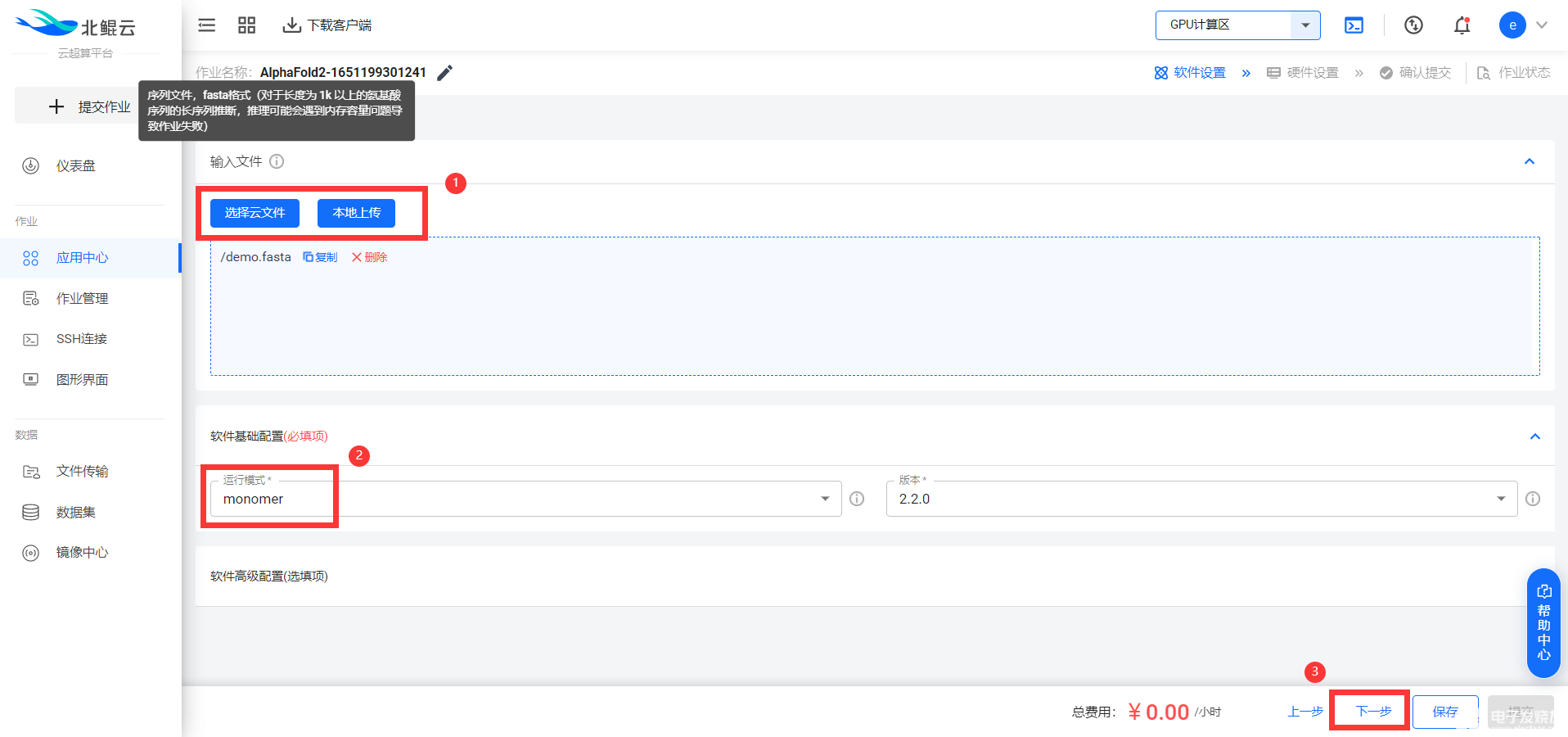
3. 填写模板参数,选择硬件配置,提交任务
上传序列文件(.fasta格式),选择运行模式(单体选择monomer,多聚体选择multimer)后即可点击下一步:
选择合适的GPU硬件配置后即可点击下一步:
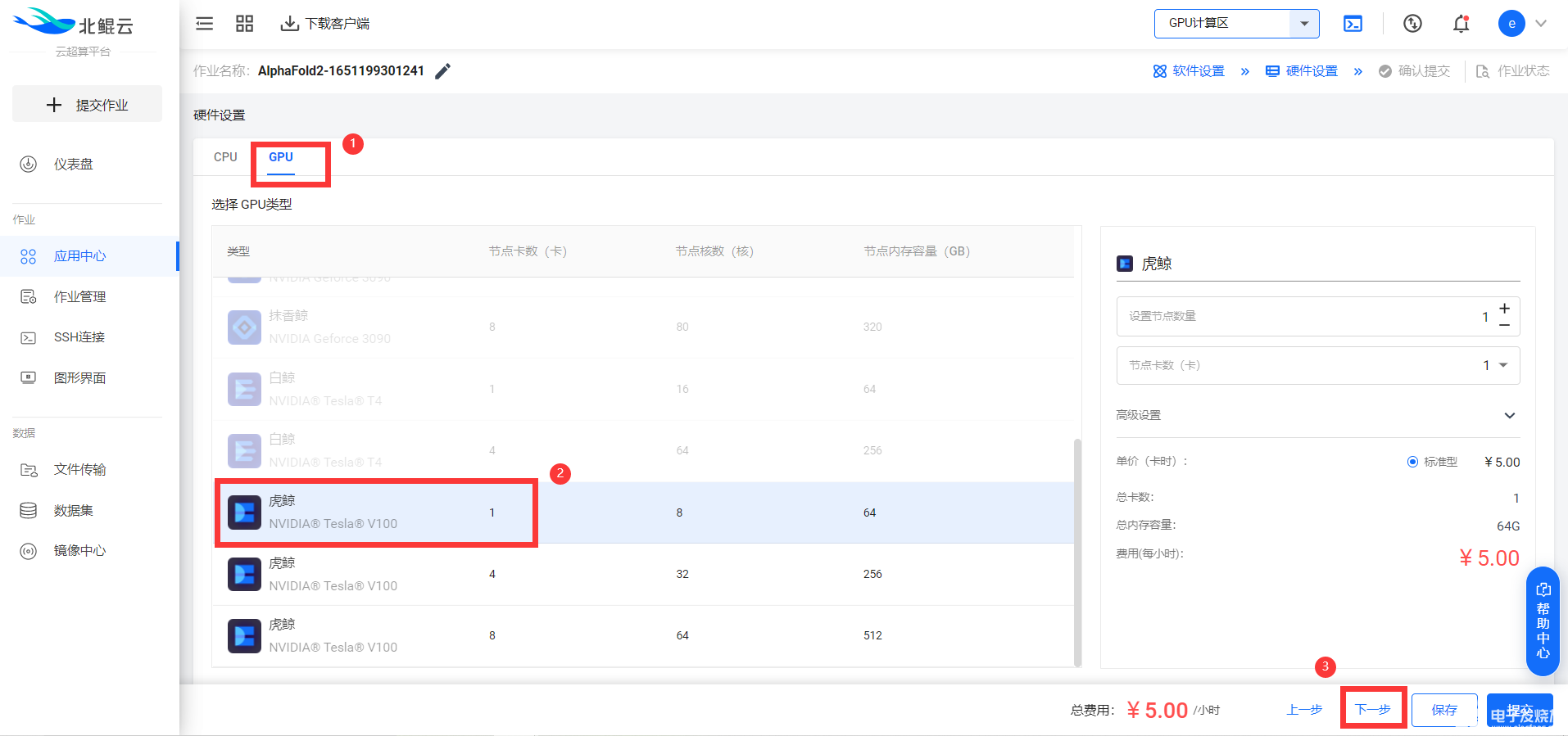
查看作业内容汇总并提交任务:
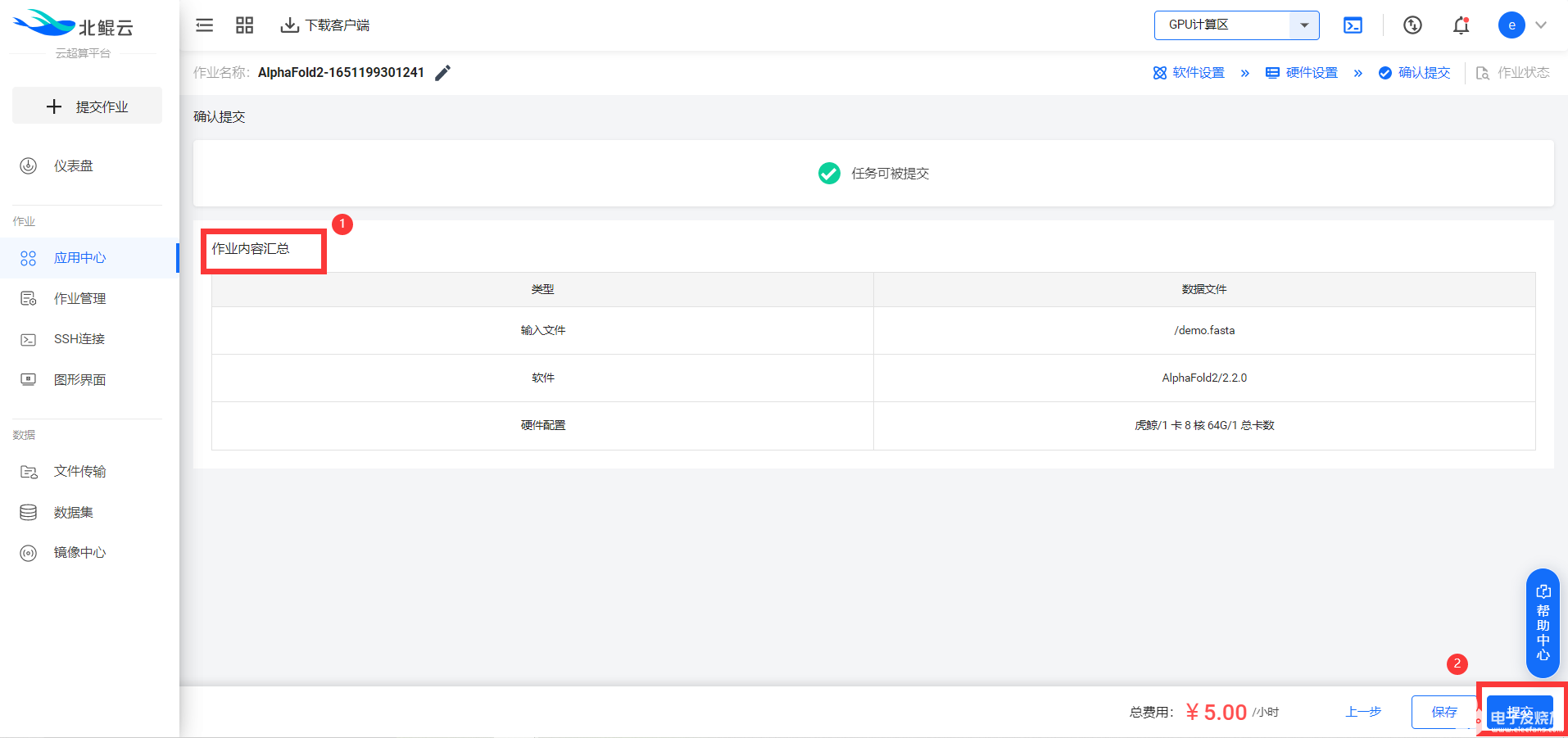
4. 查看任务详情与结果
所有通过“模板”提交的作业,都可以在左侧菜单栏“作业管理”功能中查看或者管理作业:
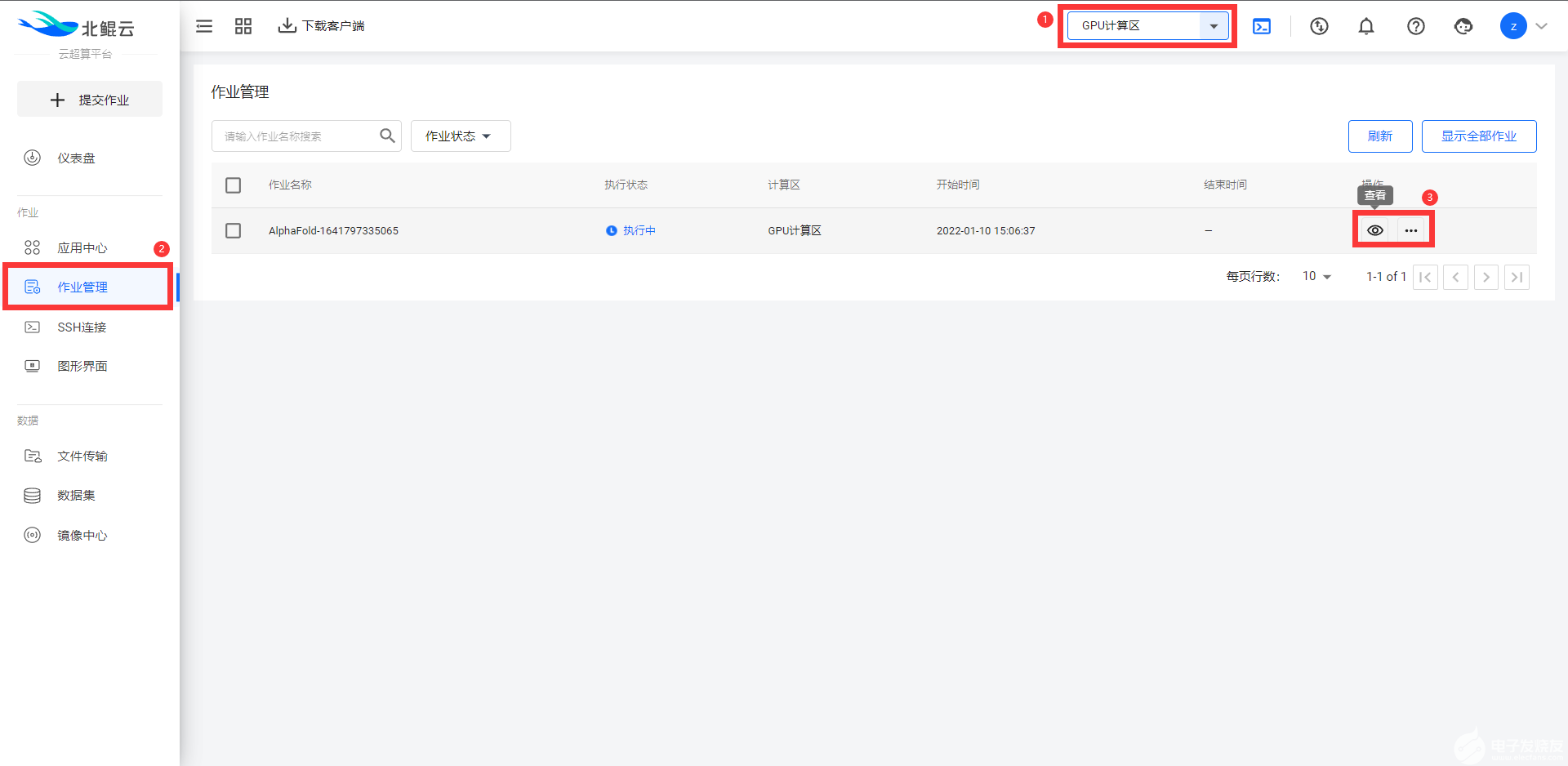
对于有Linux基础和本地硬件配置足够的人,本地使用AlphaFold2进行蛋白质预测的方法如下。
1. 配置要求
硬盘至少要3T以上,AlphaFold2训练好的模型加数据库下载下来是428 GB大小的文件,解压后需要2.2T的空间。如果你用reduced_dbs(这个是简化的数据库),那么至少也得有600 GB的硬盘空间。
12个虚拟CPU
内存85GB及以上
1个Nvidia A100 或者Nvidia V100 GPU卡
2. 下载程序需要的数据库、程序和模型
首先你得在github上面把这个AlphaFold2项目(https://github.com/deepmind/alphafold)给下载到一个本地目录,然后进入scripts这个文件夹里面,运行命令download_all_data.sh <下载目录>,程序会自动进行下载。
这个过程大概会下载438GB的文件,得等待很长时间,如果断网的话,你还得把其它的都删掉,重新下载。不建议直接运行这个主程序,可以利用多台机器分个下载。当然你也可以使用下载工具提前下载好,然后再拷贝到服务器上面去解压。
除了pdb_mmcif 这个文件之外,其它的都是可以提前下载。为什么这个文件不行?因为pdb网站并没有提供压缩的mmcif数据库文件,每个都是小文件,必须得用同步的方式把pdb服务器上面的数据库同步到本地才行,这一步建议直接在安装目录上去操作单独脚本下载,不然到时候拷贝和压缩以及解压要花大力气,这个文件夹里面有足足18万个cif文件。
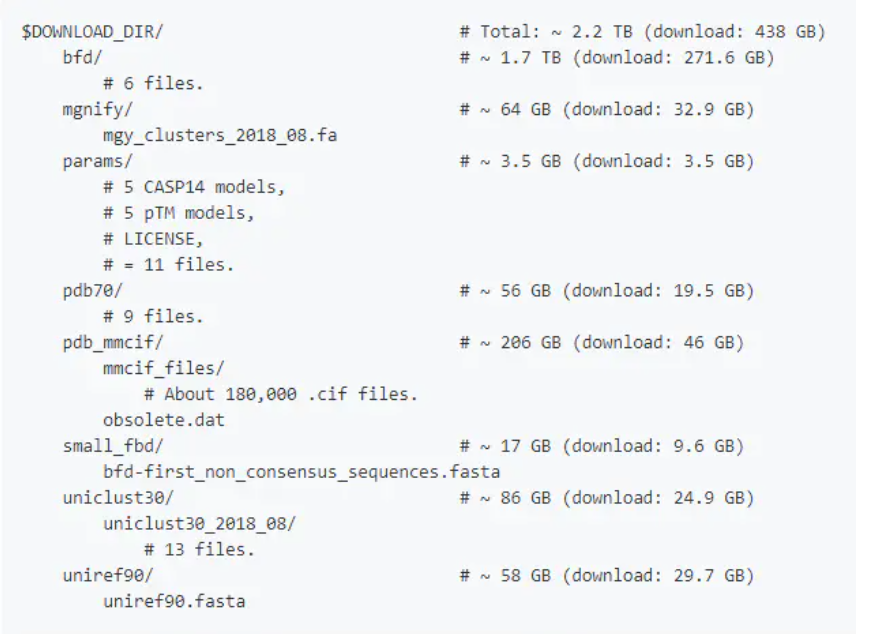
下载完成解压后关注每个文件夹文件大小和文件名是否与上面这张图中列出来的一致。
注意事项:bfd文件夹和small_bfd这两个文件夹是互斥的,大文件夹里面只留一个,bfd是完整的数据库而small_bfd是简化的数据库。如果你的磁盘不够,你就下后者,271.6 GB的bfd文件你就别下了。
3. 安装Docker和NVIDIA Container Toolkit
3.1 安装Docker
参考Docker官方教程
3.2 安装NVIDIA Container Toolkit
参考NVIDIA官方教程
3.3 测试是否安装成功
root权限运行:
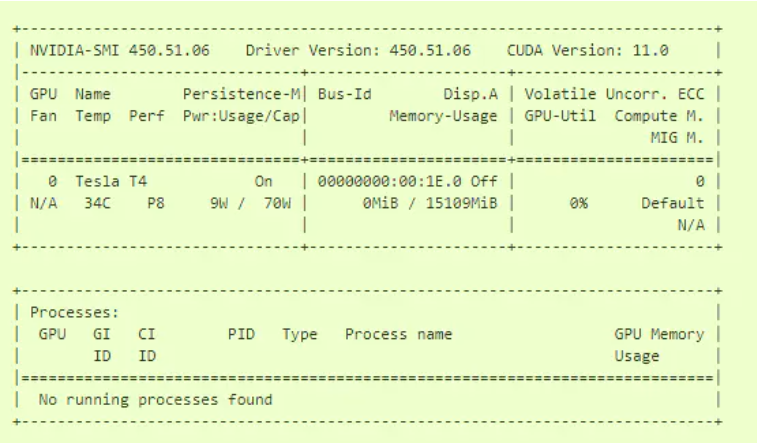
docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
如果你看到如下图的一个表格,证明你成功了。
4. 使用AlphaFold2
4.1 配置输入输出文件夹路径
首先你得配置一下输入和输出目录,打开docker文件夹下的run_docker.py脚本,然后把其中的DOWNLOAD_DIR参数改成fasta文件夹的输入目录,把output_dir后面改为输出结果的路径。
4.2 docker build
docker build -f docker/Dockerfile -t alphafold
4.3 安装pythin虚拟环境
如果你使用python3,并且机器里面有pip3,你可以直接:
pip3 install -r docker/requirements.txt
4.4 运行AlphaFold2
python3 docker/run_docker.py --fasta_paths=输入序列文件完整路径 --max_template_date=2020-05-14 --preset=[reduced_dbs、full_dbs、casp14]
fasta_paths:预测蛋白质fasta文件的文件名
max_template_date:如果你预测蛋白在pdb里面,而你不想用这个pdb做模板,你就用这个日期来限制使用该pdb做模板,这个日期应该早于这个蛋白结构的release date
preset:时间和预测质量的均衡考虑:reduced_dbs最快,但是质量最差,full_dbs中等,casp14质量最好但时间是full_dbs的八倍左右。
4.5 查看运行结果
运行结束后,在你的output_dir中会生成一系列文件,其中ranked_0到4就是AlphaFold2预测出来的分数最高的五个模型,0是最好的,可信度依次往下。
审核编辑 黄昊宇
-
人工智能
+关注
关注
1792文章
47446浏览量
239072 -
Alpha
+关注
关注
0文章
45浏览量
25660
发布评论请先 登录
相关推荐






 工商网监
工商网监
评论