 新型局部电场策略提升动力学和界面稳定性
新型局部电场策略提升动力学和界面稳定性

一、全文概要
磷基负极因其高的理论比容量和安全的锂化电位,而成为储能领域有前途的负极选择。近年来的研究表明,反应中间体(聚磷酸锂、LiPPs)在电解液中的溶解行为将导致初始库仑效率(CE)较低和不可逆容量损失较大。考虑到LiPPs的解离能小于普通锂盐,因此LiPPs更容易在电解液中解离产生带负电荷的聚磷酸盐阴离子(PPsx-)。其中,带正电荷的功能添加剂可以捕获PPsx-,减少活性物质的损失。同时,为了进一步解决动力学缓慢的问题,功能添加剂对PPsx-应该具有中等的吸附能力,从而降低反应能垒。
二、正文部分
1、成果简介
在此,天津大学孙洁教授等提出了一种新型的局部电场(LEF)策略,通过优化离子共价有机骨架(iCOFs)来抑制中间体溶解并促进反应动力学。其中,阳离子共价有机骨架诱导的LEF有效地增强了磷负极的电化学性能,多磷化物与阳离子共价有机骨架之间的强静电相互作用限制了活性材料的溶解,并调整了多磷化物的电子结构,从而加速了反应动力学。实验结果表明,阳离子共价有机骨架辅助磷负极在10.4 A g-1(8.6 C)的电流下能够提供1227.8 mAh g-1的高容量,且在 1.3 A g-1的电流下循环 500 次后,容量保持率高达 87%。因此,这一策略不仅拓宽了iCOFs在磷负极中的应用,而且激发了局部电场在电池技术中的巨大潜力。

2、研究亮点
1.本文创新性地开发了一种局部电场(LEF)策略,实现同时抑制LiPPs的溶解和促进电极反应动力学;
2.开发了iCOFs辅助石墨烯/磷负极材料的制备方法,为高性能磷基负极材料的进一步应用打下了基础。
3、图文导读
1.局域电场加速电化学反应动力学
基于已有的报道表明,磷负极锂化/脱锂过程经历了多步氧化还原过程(基于公式:P⇋LiPx⇋Li3P)。如图1a(左)所示,复杂的多相变过程直接导致了缓慢的锂化/脱锂过程。此外,中间相(LiPPs)在电解液中的溶解导致CE低,容量损失大。考虑到PPsx-的电偶极性质,本文选择了两种不同的阴离子骨架(TpPa-SO3Li)和阳离子骨架(TpEB-FSI)来探讨LEF对PPsx吸附和磷负极的反应动力学的影响。同时,将具有中性骨架的TpBD设置为对照样本,石墨也被选择作为对照样品,其作为导电碳基质,能够提高所有样品的导电性从而进行电化学性能测试。通过zeta电位测试研究了COFs的电荷性质(图1b)。TpEB-FSI的正值为35.1 mV,而石墨和TpBD的值分别为-13.8和-19.8 mV。相反,TpPa-SO3Li为-77.5 mV。在可视化实验中探讨了COFs不同电荷性质对PPsx-吸附的影响。采用化学合成法和联苯锂制备了可溶性LiPPs,LiPPs溶液的组分主要为PPsx-,如P4-、P5-和P6-及其溶剂化产物。如图1c所示,纯PPsx-溶液呈浅黄色,加入石墨、TpBD和TpPa-SO3Li后,溶液的颜色变轻,而在TpEB-FSI中,PPsx-溶液的颜色立即变得几乎无色。紫外可见(UV-vis)光谱进一步表明了TpEB-FSI具有较强的PPsx-吸附能力。
为了验证LEF对磷负极反应过程的影响,将红磷(RP)和石墨与三种材料球磨法混合,制备了不同的负极。电极材料分别记为TpEB-FSI@P/G、TpPaSO3Li@P/G、TpBD@P/G和P/G。如图1f所示,与P/G(1.05 V)、TpBD@P/G(0.86 V)和TpPa-SO3Li@P/G(0.96 V)的滞后电压相比,TpEBFSI@P/G的滞后电压最低(0.73 V),表明其低极化和优越的动力学性能。同时,充电态COFs产生的固有LEF可以通过主客电荷转移来调节PPsx-的电子结构,TpEB-FSI的阳离子骨架与PPsx-具有很强的静电吸引力,明显加速了反应动力学。相反,TpPa-SO3Li的阴离子骨架对PPsx-具有静电斥力,导致对反应动力学产生负影响,甚至比含中性共价有机框架(TpBD)的对照样品更差。通过DFT的计算研究了动力学因素,图1d分别显示了LiPPs和PPsx-在不同样品中的吸附能力。由于TpEB骨架具有独特的吸附行为,TpEB骨架在多相PPsx-上的结合能明显高于TpBD和石墨,甚至高于在LiPPs分子上的结合能。PPsx-在TpEB骨架上的强吸附可以加速锂离子的迁移,这有利于不同磷化物之间的转换动力学。根据阿伦尼乌斯方程计算出的活化能曲线(图1g),在锂化/脱锂过程中,TpEB-FSI@P/G电极的反应活化能(Ea)远低于P/G电极。这一结果表明,TpEB骨架的LEF可以加速LiPPs的反应动力学,其主要源于LEF能够改变PPsx-的电子结构。
PPsx-中的电子被TpEB的阳离子位点吸引,削弱了Li+与PPsx-之间的相互作用,使Li+易于迁移,从而具有快速反应动力学。采用电流静电间歇滴定技术(GITT)测试了Li+扩散系数(DLi),在整个放电/充电过程中,TpEB-FSI@P/G的DLi均高于P/G。其中,DLi的升高主要源于TpEB的阳离子骨架可以吸引并激活PPsx-,释放更多可移动的游离Li+。因此,TpEB-FSI可以通过LEF策略抑制LiPPs的溶解,加速LiPPs的转化,为提高磷负极的循环稳定性和倍率性能提供了一种有吸引力的方法(图1a,右)。
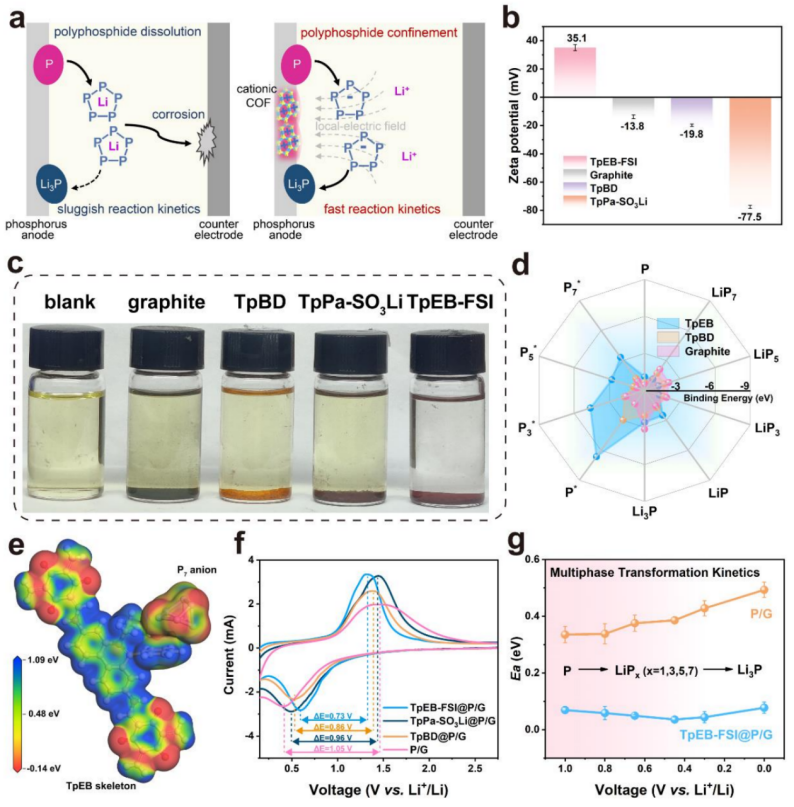
【图1】(a)磷负极常规锂化反应(左)和LiPPs在阳离子共价有机框架上的吸附和转化行为(右);(b)TpEB-FSI、TpPa-SO3Li、TpBD和石墨的Zeta电位;(c)TpEB-FSI、TpPa-SO3Li、TpBD和石墨的LiPPs可视化吸附实验;(d)LiPPs或PPsx-与TpEB、TpBD或石墨之间的结合能;(e)P7*-TpEB配合物的静电势分布;(f)TpEB-FSI@P/G、TpPa-SO3Li@P/G、TpBD@P/G 和P/G的CV曲线;(g)在放电过程中,TpEB-FSI@P/G和P/G电极在不同电压下的活化能分布。
2.iCOFs辅助石墨烯/磷负极材料的制备
本文阐述了采用一锅球磨工艺来优化磷负极的电极结构,研究了阴离子TpPa-SO3Li、中性TpBD和阳离子TpEB-FSI作为球磨过程中石墨的剥离助剂(图2a),分别命名为TpPa-SO3Li@G、TpBD@G和TpEB-FSI@G,经无保护球磨工艺获得的产品分别命名为blank@G。如图2b所示,COFs和石墨都属于高度有序的π-共轭体系,它们之间很容易形成π-π相互作用。当共价有机框架骨架携带电荷(正电荷或负电荷)时,相邻共价有机框架层之间的静电斥力驱动石墨烯薄片在剪切力下滑动。相反,由于在TpBD的中性骨架中存在较强的π-π相互作用,石墨将不倾向于分层。在球磨的强法向力作用下,TpBD@G的层结构被破坏。同时,基于TEM和AFM研究了球磨处理后的产物的形貌,blank@G和TpBD@G显示出最小的片段(图2c和d)。有趣的是,TpPa-SO3Li@G和TpEB-FSI@G表现出高质量的薄片,表明石墨被剥离成大尺寸、少层的石墨烯(图2e和f)。AFM图像进一步证明了TpPa-SO3Li@G和TpEB-FSI@G的薄层结构,其均匀厚度为~4nm(图2g)。此外,TpPa-SO3Li@G和TpEB-FSI@G具有较大的横向尺寸和超薄形貌,使其电子电导率分别为100和200 S cm-1,明显高于blank@G(8 S cm-1)和TpBD@G(10 S cm-1)。
根据以上讨论结果,采用RP、石墨和TpEB-FSI的一锅球磨工艺设计制备了TpEB-FSI@P/G复合材料。其中,TpEB-FSI的含量为RP的3 wt%,石墨/RP的质量比为3/7。如图2i所示,TpEB-FSI@P/G复合材料的ID/IG(0.86)低于P/G(2.22),说明TpEBFSI@P/G的石墨化程度相对较高。同时,P、C、N和S元素分布均匀,其中N和S属于TpEB-FSI的特征元素,说明磷和TpEB-FSI具有良好的空间组合。因此,基于TpEB-FSI对辅助石墨的剥离、抑制可溶解LiPPs的溶解、催化磷负极锂化/脱锂反应的作用,本文选择TpEB-FSI来提高磷基负极的电化学性能。
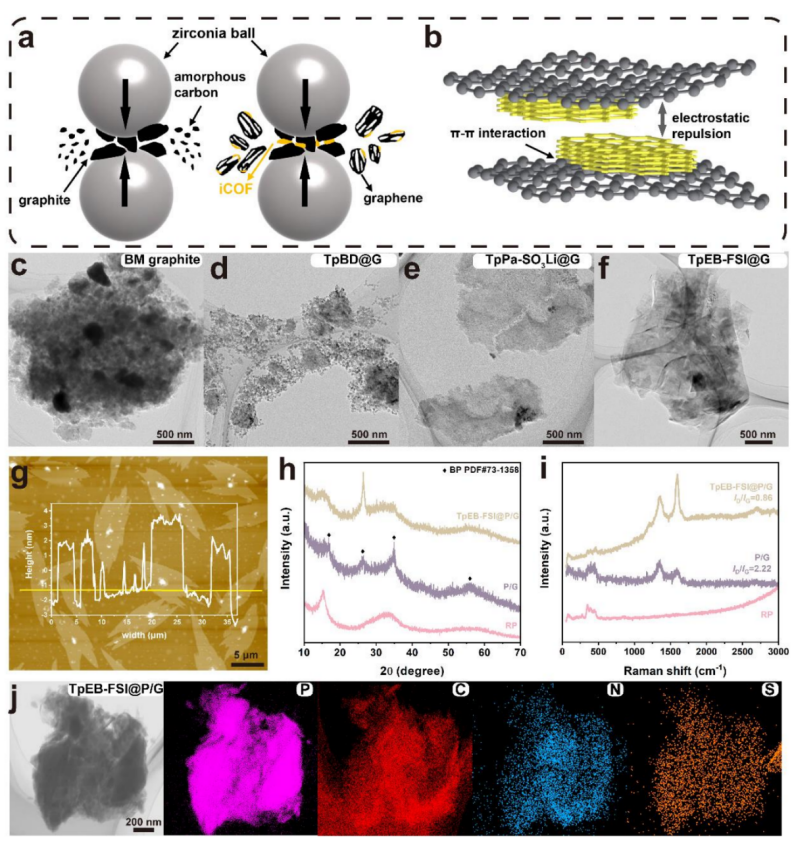
【图2】(a)非保护和iCOFs保护石墨球磨过程的示意图;(b)iCOFs球磨剥离机理示意图;(c-f)TEM图像表征;(g)AFM图像表征;(h)拉曼光谱表征;(j)TpEB-FSI@P/G的TEM图像和EDS元素映射。
3.TpEB-FSI@P/G负极的电化学性能
循环伏安法(CV)曲线如图3a所示,初始阴极扫描中在1.88 V左右的不可逆峰可归因于电解液分解引起的SEI的形成。经过初始扫描后,CV曲线的高重叠表明TpEB-FSI@P/G负极具有良好的可逆性。同时,TpEB-FSI的不同添加量(1 wt%、3 wt%和5 wt%)的TpEB-FSI@P/G负极的电化学性能不同,3 wt%的添加量为最佳选择。实验结果表明,TpEB-FSI@P/G和P/G负极在1.3 A g-1条件下,TpEB-FSI@P/G负极循环500次后表现出1626.8 mAh g-1的高容量,其保持率为87%。相比之下,P/G负极的比容量为602.0 mAh g-1,其保持率为37%。此外,TpEB-FSI@P/G负极具有更好的倍率性能,在不同电流密度下,TpEB-FSI@P/G负极的过电位(ΔEp)明显低于P/G负极,表明具有超快反应动力学。在高电流密度为5.20 A g-1时,TpEB-FSI@P/G负极也表现出稳定的长时间循环性能(图3g)。结果表明,将TpEB-FSI引入磷负极纳米结构具有良好的锂存储性能。
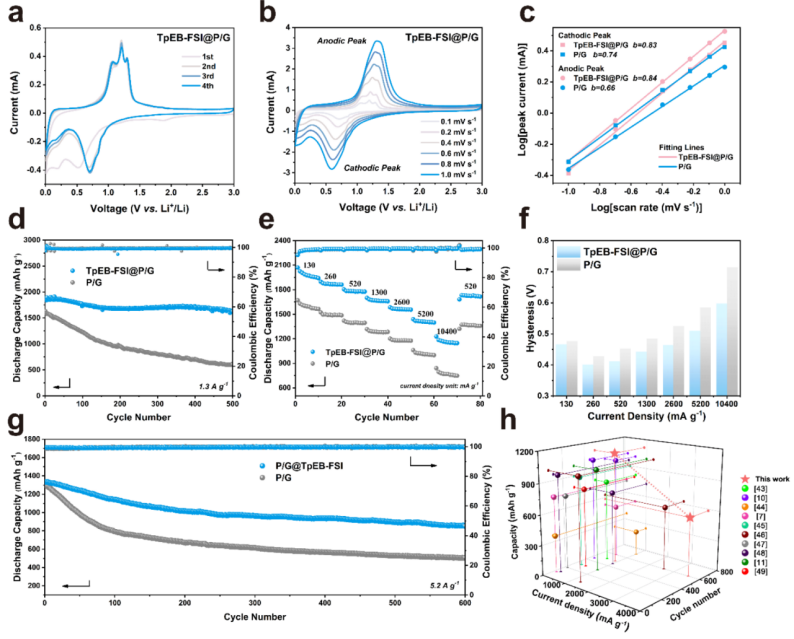
【图3】(a)TpEB-FSI@P/G的CV曲线;(b)TpEBFSI@P/G在不同扫速下的CV曲线;(c)每个氧化还原峰对应的图;(d)循环性能;(e)倍率性能;(f,g)过电位和长循环测试;(h)TpEB-FSI@P/G与其他关于P基负极的电化学性能比较。
4. 局部电场调控电极界面
当电极与电解液接触时,在两相边界处产生界面,导致电极与电解液之间的热力学电位存在间隙,该界面的组成和性质对稳定循环性能起着关键作用。然而,在充放电循环过程中,磷的体积变化使SEI不稳定,新暴露的电极表面将继续与电解液发生反应,导致电极粉碎和循环性能恶化,稳定、坚韧的SEI可以延长电池的使用寿命。本文将含有阳离子骨架的TpEB-FSI与含有中性骨架的TpBD进行了比较,以强调其在调节界面特性方面的独特LEF效应(图4a)。如图4b所示,与TpBD@P/G相比,TpEB-FSI@P/G表现出更好的循环稳定性,且利用初始充放电电压曲线来探索SEI的形成过程(图4c)。TpEBFSI@P/G的初始CE为68.9%,低于TpBD@P/G的初始CE(87.7%),低CE表明在初始放电过程中发生了更多的不可逆反应。在存在TpEB-FSI@P/G的情况下,由于界面上的氟离子和阴离子的分解,在1.2 V以上产生不可逆容量。相比之下,在TpBD@P/G中没有观察到不可逆的分解反应。
同时,采用AFM测量了循环后TpEB-FSI@P/G和TpBD@P/G负极的形貌和杨氏模量(图4f和g)。TpEB-FSI@P/G负极呈现出整体而光滑的表面,而TpBD@P/G负极呈现出破碎而粗糙的表面。此外,TpEB-FSI@P/G SEI的杨氏模量明显高于TpBD@P/G SEI,使得TpEBFSI@P/G负极的界面坚硬,能够维持电极结构,缓解体积膨胀的影响。此外,TpEB-FSI@P/G的大尺寸、少层石墨烯结构可以作为缓冲层来容纳体积膨胀。
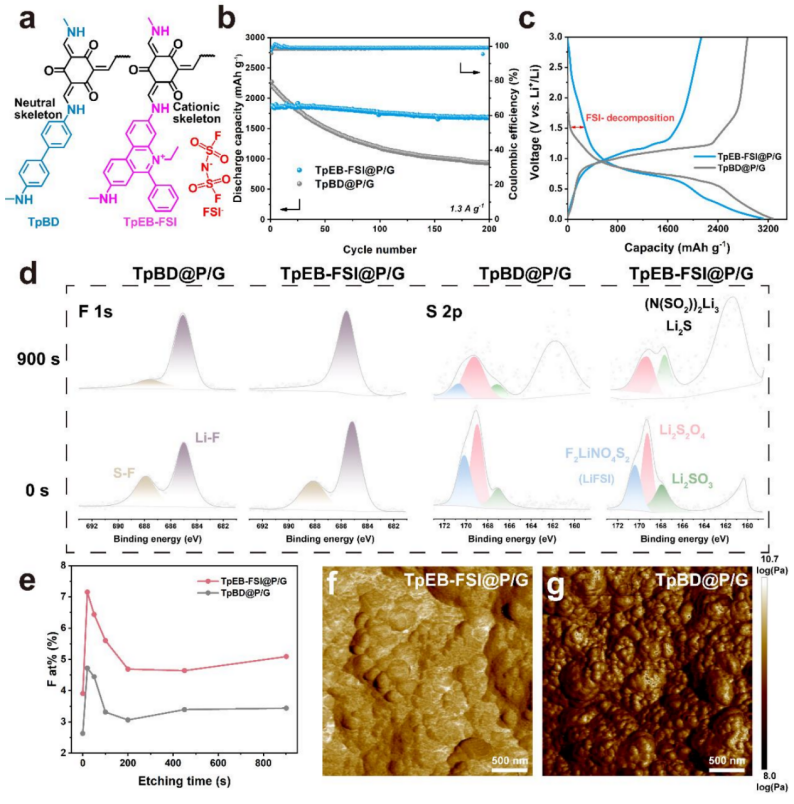
【图4】(a)TpBD和TpEB-FSI的分子结构;(b)TpEBFSI@P/G和TpBD@P/G负极在1.3 A g-1条件下的循环性能;(c)在0.13 A g-1条件下,TpEBFSI@P/G和TpBD@P/G负极的充放电电压曲线;(d)不同蚀刻时间下循环后磷负极表面的高分辨率F 1s和S 2p XPS光谱;(e)不同蚀刻时间下的F含量;(f,g)TpEB-FSI@P/G和 TpBD@P/G负极的杨氏模量。
5.磷负极局部电场特性的探讨
低电导率、不稳定的SEI、可溶性的中间体和有限的反应动力学阻碍了磷负极性能的提高,本文提出的阳离子共价有机框架为解决上述问题创造了有效的LEF效应策略。LEF效应的优势主要体现在以下几个方面(图5):(i)阳离子共价有机框架作为剥离辅助剂,促进大尺寸石墨烯的形成,为快速电化学反应提供了连续的电子转移途径;(ii)阳离子共价有机框架纳米通道中的阳离子位点产生了LEF效应,诱导锂盐分解反应形成稳定、坚韧的富含LiF的SEI,从而有效地提高了磷负极的界面稳定性;(iii)LEF还可以限制可溶性LiPPs的溶解,改变PPsx-的电子结构,从而加速反应动力学。
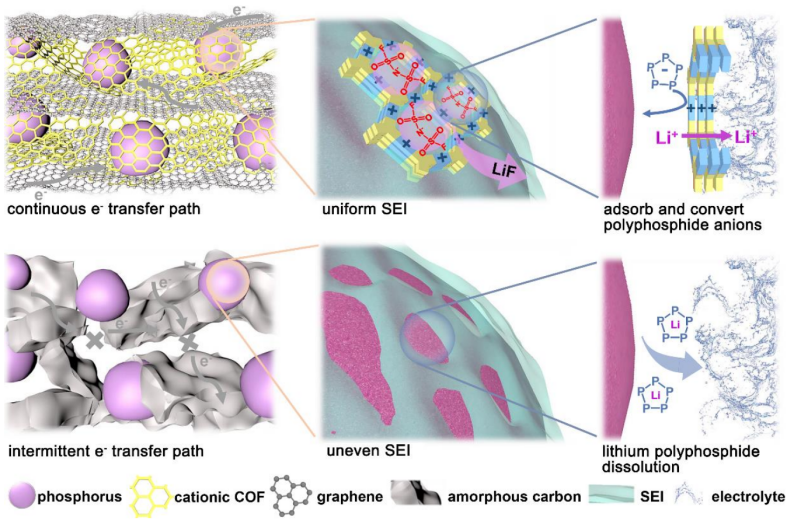
【图5】
4、总结与展望
本文针对新发现的LiPPs溶解和磷负极动力学缓慢等问题,构建了一种新的具有调控LEF的阳离子共价有机框架。PPsx-和阳离子共价有机框架之间的静电吸引降低了LiPPs在电解液中的溶解,从而缓解了活性物质的损失。具体来说,受限的PPsx-受到阳离子共价有机框架骨架中LEF的影响,进一步释放更多移动的自由Li+,加速锂化/脱化动力学。同时,LEF还可以诱导产生稳定和坚韧的SEI,以实现长循环的稳定性。此外,本文开发了一种新型的iCOFs辅助球磨技术,以制备用于磷负极的高质量石墨烯,该石墨烯展现出优异的电导率(~200 S cm-1)。因此,本研究为构建电池系统的LEF策略开辟了新的途径,其也适用于存在溶解问题(如S和LiNi1-x-yCoxMnyO2正极)和界面不稳定(如硅和碱金属负极)的各种电极材料。审核编辑:郭婷
-
电流
+关注
关注
40文章
7228浏览量
141617 -
电解液
+关注
关注
10文章
881浏览量
23859
原文标题:天津大学孙洁Adv. Mater.:聚焦磷基负极快充,局部电场提升动力学和界面稳定性
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
Altair Radioss:瞬态动力学仿真专家?
微电网稳定性理论在实际应用中面临哪些挑战

微电网稳定性分析:小干扰稳定与暂态稳定关键技术

多物理场耦合动力学:机电液耦合下无人机起落架伺服系统解耦控制与动态响应优化研究

Neway微波的稳定性优势
CW32 MCU在高频率运行下的系统稳定性的提升方案
界面层创新:全固态钠电池稳定性实现突破性提升
功率放大器赋能:压电双晶片动力学研究的突破之旅
如何保证合金电阻的稳定性与精度?

如何利用X-Ray技术提升锂电池安全性与稳定性
三坐标如何实现测量稳定性的提升




评论