 调控几何位点占据实现尖晶石钴氧化物高效析氧反应
调控几何位点占据实现尖晶石钴氧化物高效析氧反应

01
导读
化石燃料的日益枯竭引起了人们对新型能源转换技术如电解水技术的日益关注。然而较高过电位和缓慢动力学严重阻碍了析氧反应(OER)的发生。具有可控的A、B几何位点的AB2O4型尖晶石氧化物,由于其低成本、高元素丰度和可调控的电子行为,是一类有前途的析氧反应催化剂。
然而,开发在碱性介质中具有优异OER性能的新型尖晶石氧化物仍然是具有挑战的。许多基于过渡金属的催化剂在OER过程中存在不可逆的结构重构,并在表面形成新的物种,这种重构的表面是析氧反应的真正活性位点。掺入过渡金属可以引入更多的氧缺陷以促进自重构。然而,桥接取代阳离子的初始几何位点占位并实现高OER活性/稳定表面的可变重构仍较难实现。
02
成果背景
近期,Angew. Chem. Int. Ed.期刊上发表了一篇题为“Balancing Activity and Stability in Spinel Cobalt Oxides through Geometrical Sites Occupation towards Efficient Electrocatalytic Oxygen Evolution”的文章。该工作在不破坏(Co)tet(Co2)octO4的正常尖晶石结构情况下制备了一类Ni/Mn取代的(Co)tet(Co2)octO4(其中"tet"表示四面体位点,"oct"表示八面体位点)尖晶石氧化物纳米片(NSs),其具有优异的析氧活性,在10 mA cm−2时过电位仅为281.6 mV。
03 关键创新
镍/锰的取代改变了CoCo2O4的正常表面状态,使其从过氧状态转变到缺氧状态,引发非耦合质子-电子转移从而重建更稳定的表面。
04
核心内容解读
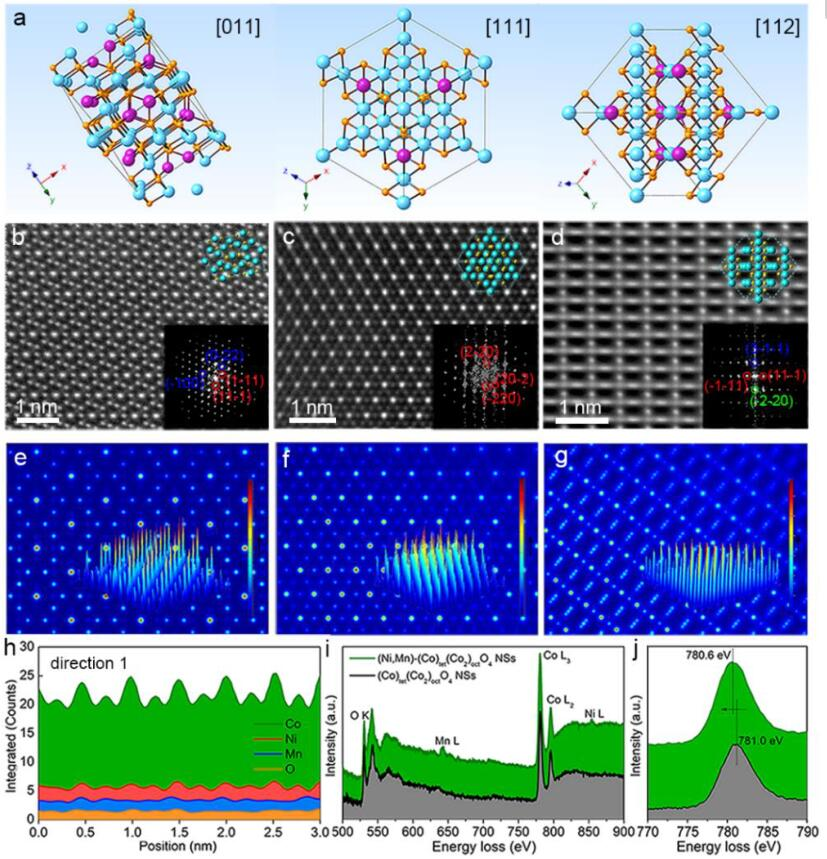
图1 (a)沿[011]、[111]和[112]方向的晶胞透视图;(b-d)分别在[011]、[111]和[112]方向观察到的(Ni,Mn)-(Co)tet(Co2)octO4NSs的HAADF-STEM图像;(e-g)TEM/STEM模拟包软件(QSTEM)模拟的STEM理论图像,分别沿[011]、[111]和[112]方向投影;(h)(Ni,Mn)-(Co)tet(Co2)octO4NSs的原子分辨率EDS线扫描;(Co)tet(Co2)octO4NSs和(Ni,Mn)-(Co)tet(Co2)octO4NS的(i)完整和(j)放大EELS光谱。
尖晶石氧化物的电子结构受过渡金属阳离子在八面体或四面体位点的空间分布影响很大。CoCo2O4沿[011]、[111]或[112]投射的四面体A位和八面体B位所占的有序金属原子比为1:2(图1a)。三个方向的电子衍射图案也验证了杂原子Ni/Mn被成功地替代到(Co)tet(Co2)octO4晶格中,但(Ni,Mn)(Co)tet(Co2)octO4NSs的基本结构仍为尖晶石结构(图1b-d的插图)。
同时,(Ni、Mn)-(Co)tet(Co2)octO4NSs的原子分辨EDS元素面扫描图和对应的三个方向的EDS线扫描图显示了Ni和Co相同的排列和起伏形式,揭示了Ni具有与Co相似的几何位点占据(图1h)。根据(Co)tet(Co2)octO4NSs和(Ni、Mn)-(Co)tet(Co2)octO4NSs的EELS光谱信息,Ni/Mn取代(图1i,j)可以降低Co的价态。
由于Ni/Mn掺杂原子的存在,O 2p和Co 3d的杂化轨道也发生了变化,表明(Ni,Mn)-(Co)tet(Co2)octO4NSs的表面氧状态发生了改变。
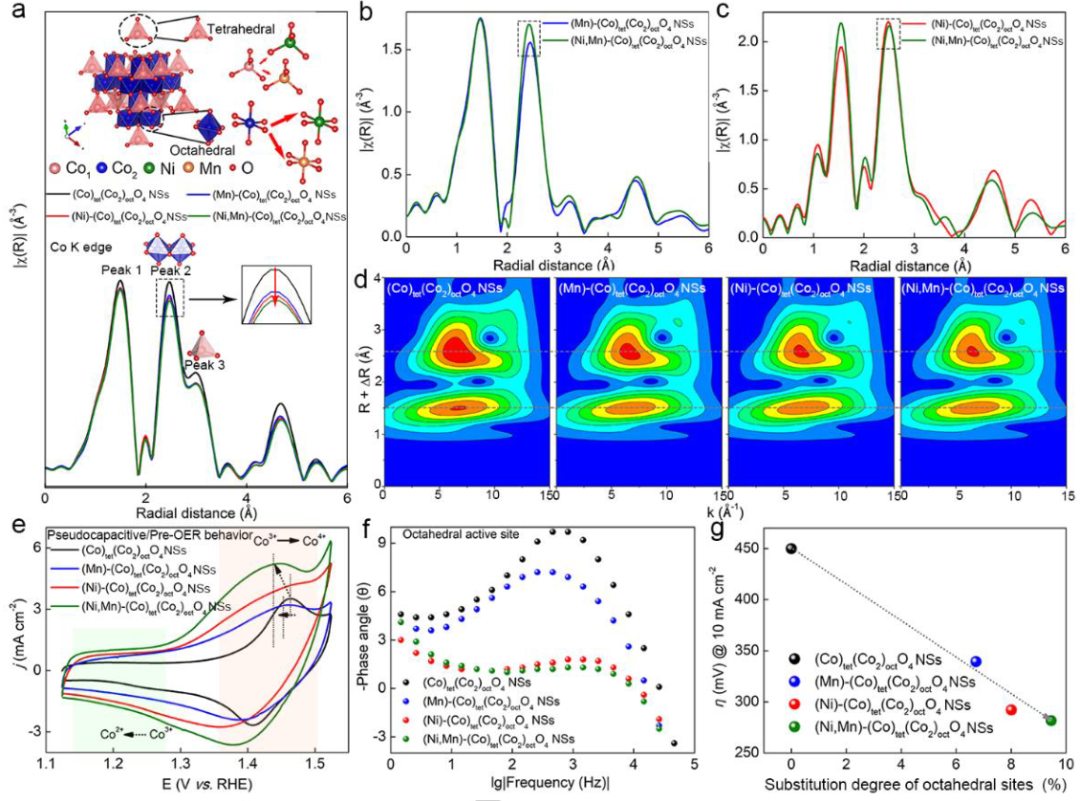
图2(M)-(Co)tet(Co2)octO4(M=Ni/Mn)NSs的(a)Co K-边,(b)Mn K-边和(c)Ni K-边的傅里叶变换(FT) k3χ(R),(d)k3加权Co K边EXAFS信号的小波变换(WT);(e)在O2饱和的1.0 M KOH中的CV曲线;(f)在1.524V(与RHE)下相角作为频率的函数;(g)η@10 mA cm-2(mV vs. RHE)与八面体位点的取代度(%)之间的线性关系。
(M)-(Co)Tet(Co2)octO4(M=Ni/Mn)NSs的Co K边EXAFS光谱在0.0-4.0 Å内有的三个峰,分别对应于Co-O(1.0-2.0Å,峰1)、Cooct-Cooct(约2.5Å,峰2)和Cotet-Cooct/Cotet-Cotet(约3.0Å,峰3)。峰1强度减弱表明(Co)Tet(Co2)octO4NSs的钴-氧配位数在引入Ni/Mn后变低,CoCo2O4的表面状态转变为缺氧状态。
Mn K-边和Ni K-边处的EXAFS曲线显示Mnoct-Moct和Nioct-Moct的强度明显高于MnTet-Mtet/Moct和Nitet-Mtet/Moct的强度,表明引入的Mn和Ni倾向于占据几何八面体位置。OER过程中可以观察到两对氧化还原峰,分别属于Co2+/Co3+和Co3+/Co4+的氧化还原对(图2e)。
具有最大Co3+/Co4+氧化峰面积的(Ni,Mn)-(Co)Tet(Co2)octO4NSs显示出从1.463到1.437 V的显著偏移,说明(Ni,Mn)-(Co)Tet(Co2)octO4NSs更容易被预氧化和去质子化。
图2g表明,具有较高取代度、较大Co2+/Co3+比、较低价态钴和较小八面体位置eg占据的(Ni,Mn)-(Co)tet(Co2)octO4NSs对OER更有活性。规律性相关揭示了Nioct和Mnoct对OER活动的重要影响。
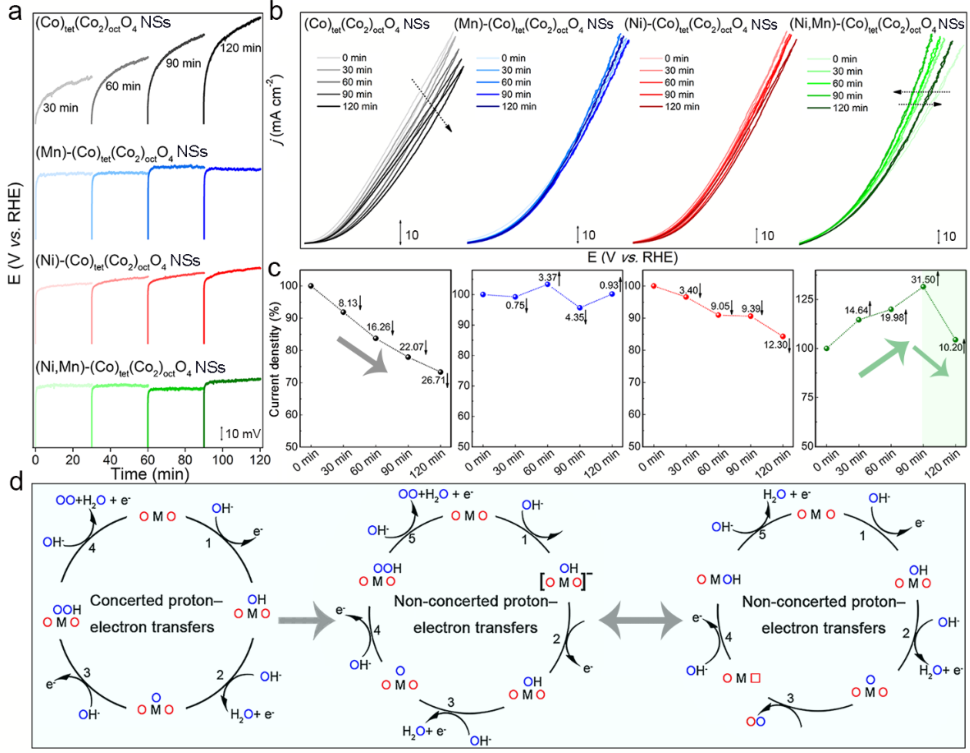
图3(M)-(Co)tet(Co2)octO4(M=Ni/Mn) NSs(a)在10 mA cm-2下对OER的计时电位响应;(b)在不同时间的CP测量后的CV曲线;(c)电流密度变化;(d)传统的协同质子-电子转移机制(左);一种非协同质子-电子转移机制(中);由晶格氧的氧化还原引发的非协同质子-电子转移路径(右)。
使用连续计时电位法(CP)-循环伏安法(CV),测试了催化剂的稳定性和活化趋势。经杂原子取代的氧化物催化剂,比(Co)tet(Co2)octO4具有更好的稳定性(图3a)。在CP测量中,在(Ni,Mn)-(Co)tet(Co2)octO4NSs上出现了约31.50%的显著活性提升(图3b,c)。
Ni和Mn占据的最大八面体位置可能会将传统的协同质子-电子转移机理转换为非协同质子-电子转移过程(图3d)。图3b、c中120 min-CV降低10.20%证实了在动态优化后,(Ni,Mn)-(Co)tet(Co2)octO4NSs的活性回到初始值并保持动态稳定性。
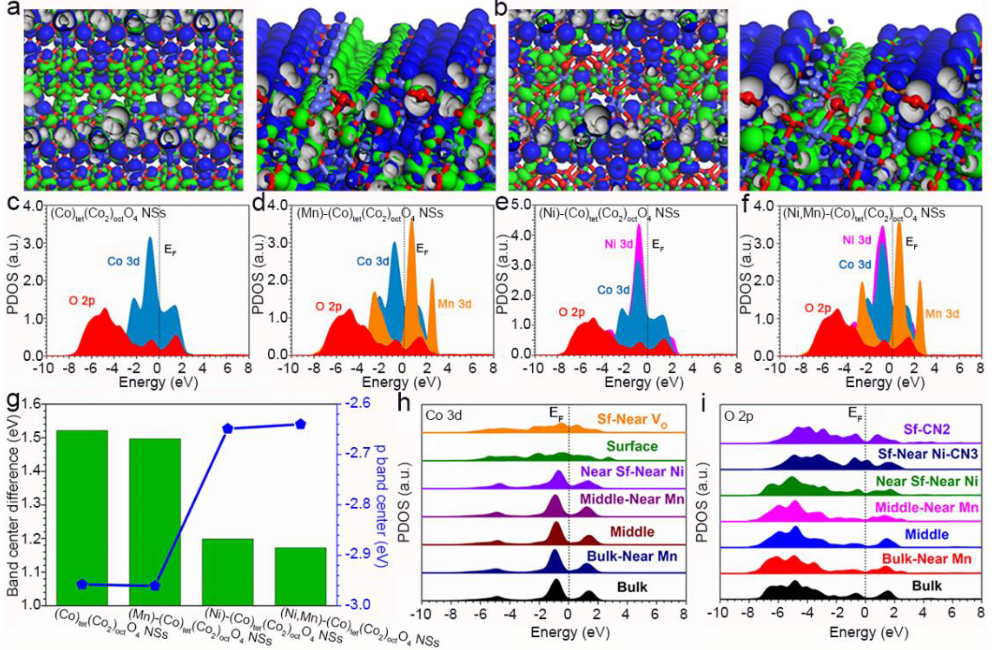
图4(a)(Co)tet(Co2)octO4和(b) (Ni,Mn)-(Co)tet(Co2)octO4的费米级附近电子分布的三维等高线图;(c)(Co)tet(Co2)octO4,(d)(Ni)-(Co)tet(Co2)octO4,(e)(Mn)-(Co)tet(Co2)octO4,和(f)(Ni,Mn)-(Co)tet(Co2)octO4的PDOS;(g)不同结构的d带中心和p带中心;(Ni,Mn)-(Co)tet(Co2)octO4中(h)Co 3d的PDOS和(i)O 2p的PDOS。
作者采用密度泛函理论研究了(Ni,Mn)-(Co)Tet(Co2)octO4的OER性能提升原因。引入Ni和Mn原子不会引起费米能级(EF)附近的电子分布发生显著变化。Ni和Mn位点的结合没有引起结构的坍塌,表明构建的结构模型是稳定的。图4g总结了(M)-(Co)tet(Co2)octO4(M=Ni/Mn)结构中O的p带中心和Co的d带中心。
(Co)tet(Co2)octO4的O p带中心和Co d带中心之间显示出最大的势垒,其活性最低。而(Ni)-(Co)tet(Co2)octO4和(Ni,Mn)-(Co)tet(Co2)octO4提升了O p带中心,导致d带中心和p带中心之间的差异减小。此外,在(Ni,Mn)-(Co)tet(Co2)octO4中p-d带的最小差异支持最高的电子转移效率,从而提高OER性能。
Co位置的几何位置依赖性PDOS表明,Ni和Mn位置显示了对附近的Co 3d轨道的相反影响,其中Ni引起上移,而Mn导致下移(图4h)。八面体中心的低配位Co位显示出有效的电子转移能力。而O 2p没有发生显著变化(图4i)。然而,O 2p轨道被在表面的Ni位点激活,进一步促进了碱性OER的位点间电子转移。
从电子结构的角度来看,(Ni,Mn)-(Co)tet(Co2)octO4的最佳OER性能来源于高电活性和稳定性之间的最佳平衡。
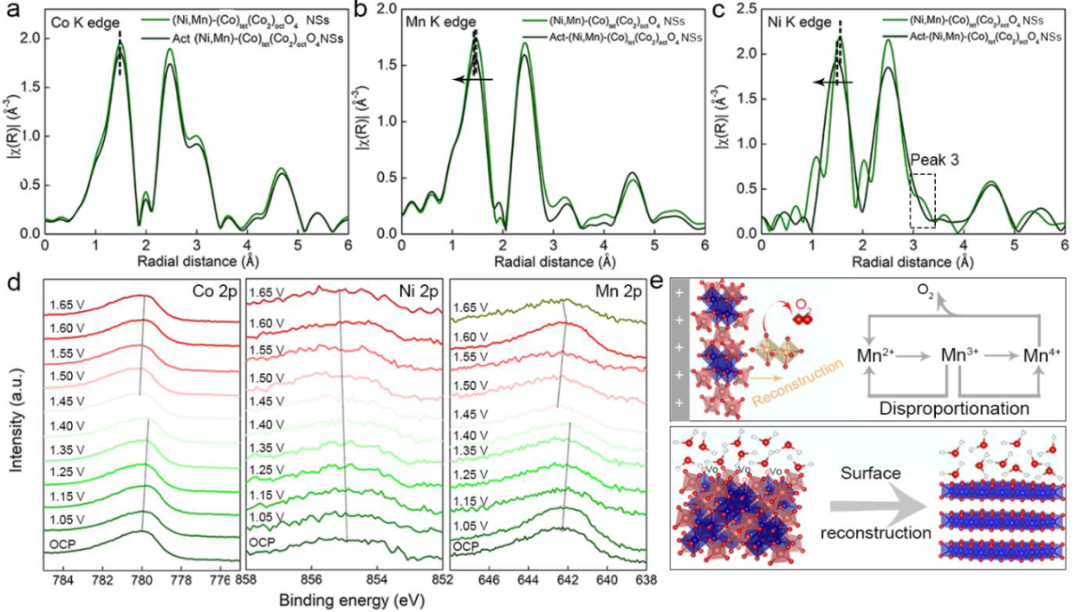
图5在OER激活过程前后,(Ni,Mn)-(Co)tet(Co2)octO4的(a)Co K-边,(b)Mn K-边和(c)Ni K-边的FT-EXAFS图谱;(d)准原位XPSCo 2p,Ni 2p,和Mn 2p谱图;(e)歧化过程和表面重构的示意图。
(Ni,Mn)-(Co)Tet(Co2)octO4和Act-(Ni,Mn)(Co)Tet(Co2)octO4NSs在Co K边缘、Mn K边缘和Ni K边缘的EXAFS曲线如图5a-c所示。三个峰分别表示平均TM-O键、TMoct-TMoct键和TMtet-TMtet/TMoct键。
在Ni K边缘和Mn K边缘可以观察到轻微压缩的M-O键,表明部分表面氧化。然而,Co-O键的长度几乎保持不变,这表明Ni和Mn的存在使表面Co更加稳定。三种元素的TMoct-TMoct键和TMtet-TMtet/TMoct键强度的降低反映了OER活化后不同的金属位置占据条件。
与(Ni,Mn)(Co)tet(Co2)octO4的曲线相比,在电化学活化后,Ni K边缘峰3消失,说明Nitet迁移到未占据的八面体位置或在此过程中浸出到电解液中。准原位XPS结果(图5d)显示,随着电压增加,Co的价态逐渐上升,并且在1.45 V处出现明显的相移。
因此,推测表面重构主要发生在1.40 V和1.45 V之间。在Ni 2p XPS谱图中观察到更高结合能的移动,表明在氧化过程中Ni的价态增加。这种具有还原离子半径的氧化Ni更倾向于占据几何八面体位置,并促进表面重构。
Mn 2p的结合能随着施加的电势从开路电势(OCP)到1.60 V发生降低,在表面相变之后,在1.45 V时急剧变化,并且在1.65 V增加。这种现象可能是表面重排和Mnoct在重构表面上的动态平衡过程的结果,使得(Ni,Mn)-(Co)Tet(Co2)octO4NSs成为具有稳定的自组装表面的催化剂。
05
成果启示
该工作合成了一类(M)(Co)Tet(Co2)octO4(M=Ni/Mn)NSs,其中(Ni,Mn)(Co)tet(Co2)octO4NSs通过调节取代元素的几何八面体位置占据而产生优异的OER电活性。Ni/Mn杂原子掺杂改变了CoCo2O4的表面状态,使其从过氧状态转变到缺氧状态。密度泛函理论计算表明,镍和锰位点可以通过有效的位点间电子转移提高活性。在表面重构之后,此催化剂在100 mA cm-2下具有100 h的优异稳定性。本文为设计理想的电催化剂提供了关键的指导。
审核编辑:刘清
-
电解液
+关注
关注
10文章
881浏览量
23857 -
OCP
+关注
关注
0文章
85浏览量
17125 -
EDS
+关注
关注
0文章
105浏览量
12339
原文标题:Angew:调控几何位点占据实现尖晶石钴氧化物高效析氧反应
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
四探针方阻仪高精度表征:铟镓锌氧化物/银/铟镓锌氧化物多层膜的光电性能

君耀40D系列金属氧化物压敏电阻技术参数及选型应用分析

四探针法 | 测定氧化物离子导体的电阻率
氧化物正极的化学密码:电子构型、化学键合与化学反应性如何主宰电池性能

NiOₓ表面电化学活性位点的高效钝化及其在钙钛矿电池中的应用

TDK SIOV-S14K系列金属氧化物压敏电阻:高性能与可靠性的完美结合
TDK SIOV-S10K***K11金属氧化物压敏电阻:小尺寸大作用
TDK SIOV-S07K系列金属氧化物压敏电阻:高性能与可靠性的完美结合
能量调控的精巧使者:ZK68N80T MOSFET的特性与价值

固体氧化物燃料电池/混合电推进系统:热力学耦合机制与能量梯级利用优化

柳江沙塘段网箱养殖水质智能调控:凯米斯科技微系统赋能生态养殖升级

ROBOT之鼻金属氧化物半导体气体传感器静电浪涌防护技术

连续焦耳加热赋能三元协同催化剂,高效水处理方案来袭




评论