 如何提高HEAs催化剂的催化活性和优选设计研究
如何提高HEAs催化剂的催化活性和优选设计研究

01、导读
当今世界面临着能源需求增长与遏制全球变暖两大挑战,因而需要开发化石燃料的替代品。其中,基于氧还原反应(ORR)的氢燃料电池扮演了重要角色。但ORR的动力学缓慢,必须引入催化。常用催化剂(如Pt)成本高昂,开发活性更高、成本更低的催化剂至关重要。高熵合金(HEAs)由于在连续组成空间里不定的、近乎无穷的组分数而被视为极具潜力的开发新电催化剂的平台,如何提高HEAs的催化活性也成为新兴的重要课题。
02、成果背景
近期,Advanced Energy Materials发表了题为“Following paths of maximum catalytic activity in the composition space of high-entropy alloys”的文章。该工作以五元HEAs(Ag-Ir-Pd-Pt-Ru)为模型进一步探究了ORR反应活性与HEAs组分的关系。结合机器学习与修正的微动弹性带(NEB)方法的模拟结果表明ORR反应活性最大值与“山脊线”有关。该工作为提高已知组分HEAs催化剂的催化活性和优选设计新催化剂提供了新的思路。
03、关键创新
(1)改进NEB算法以实现降低预测最大催化活性路径的计算量;
(2)沿着催化活性山脊线到组成空间的边缘,进行元素替换并进行模拟。从而提高催化活性并优选设计新催化剂。
04、核心内容解读
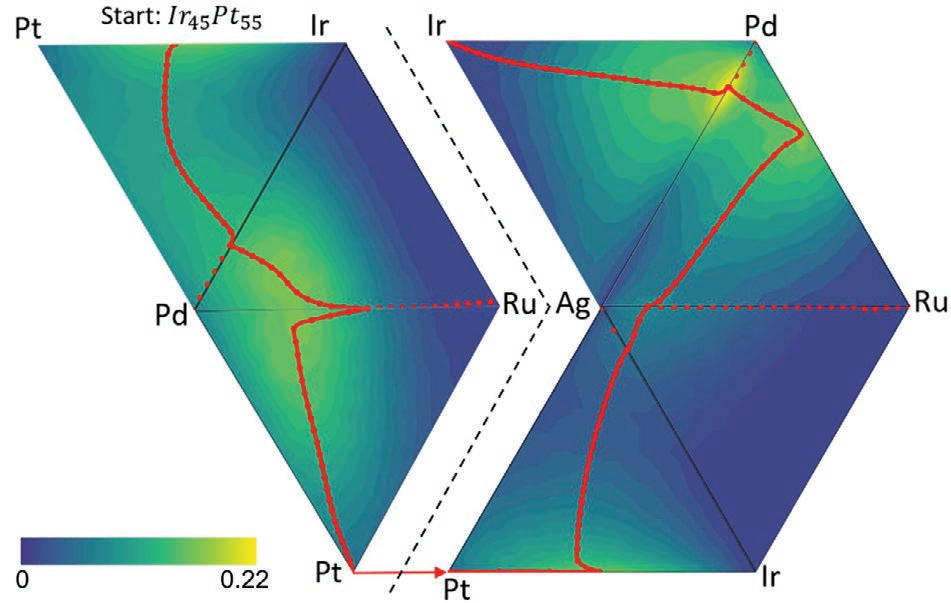
图1三元亚空间中自Ir45Pd55开始的元素取代示意图。
图1中选取一种已知的具有催化活性的双金属合金Ir45Pd55为起始点,随机选取Pd以构成初始的三元空间,并将其作为NEB方法的计算终点。图中7个三角形代表NEB方法经历的7个三元亚空间。模拟结果成功找到了全局最优值Ag17Pd83,并且找出了各三元组分的局部最优值。由此可推出所有最大值确实沿山脊线相连,且替换边缘元素是一种提高催化活性的有效方法。这一推论也在五元合金Ag-Ir-Pd-Pt-Ru中得以验证(图2a),且在其经历的最大值中有数个跟之前的报道相符。
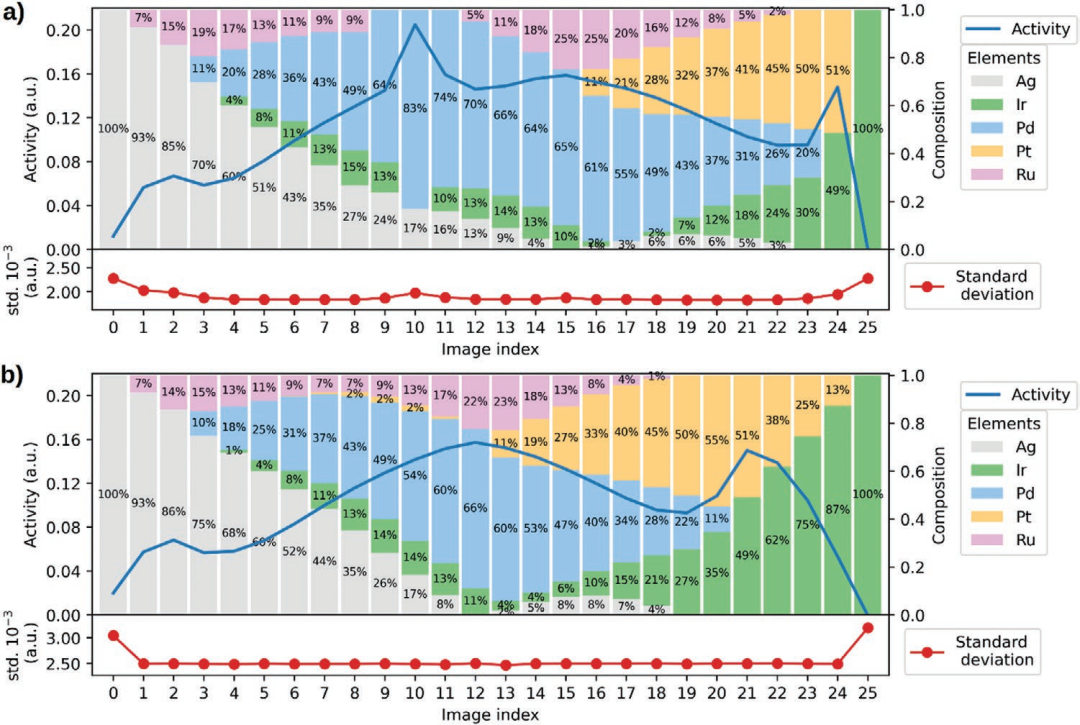
图2使用GPR模型训练的经典NEB算法(a)和机器学习NEB算法(ML NEB)(b)的Ag和Ir之间收敛NEB路径的活性高度分布(5%原子含量梯度)。
经典NEB算法需要进行大量模拟工作,而ML NEB算法则致力于对每次迭代中最不确定的值进行抽样以减少工作量。图3比较了Ag-Ir-Pd三元组分空间中2种NEB算法的路径。二者路径基本相同,但ML NEB算法只用33次估算,而在5%原子含量梯度下总样品达231个,可见简化效果极为明显。但当组分空间中含元素更多时,ML NEB算法则会略去某些最优值。如在图2b中,图2a中的全局最优值Ag17Pd83被忽略。此现象的一个可能原因是ML NEB算法忽略了某些梯度信息。总体来说,随着组分空间中元素增加,使用ML NEB算法对整个空间进行精确预测的难度越来越大。
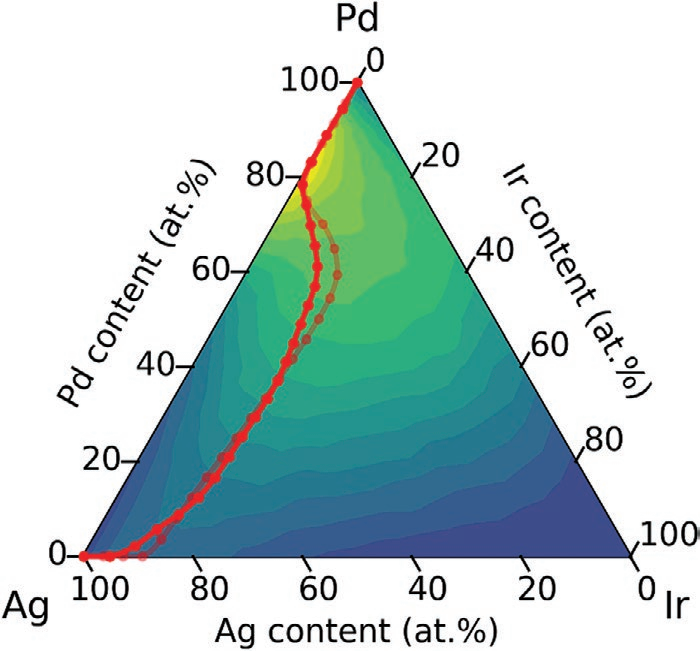
图3经典NEB算法与ML NEB算法的对比。
对于原子体系的ML NEB算法,GPR会使用梯度信息以增加预测精度。但在此工作中表面并非能量表面,因而梯度信息不能直接从对催化活性的模拟中提取。而GPR中的梯度信息会实现更快和更精确的收敛路径。Ag17Pd83被忽略也与组分空间中山脊线的形状有关。在图2a中,朝着Ag17Pd83去的山脊线像“死胡同”一样而非连续的,这是因为Ag17Pd83前后的不同摩尔比似乎代表了同一路径,可以形象地认为ML NEB算法找到的路径在Ag17Pd83面前“转了个身”。
为尝试理解组分空间中山脊线的本质,作者又开发了山脊线检测算法,这种算法能判断一个给定的点是否在训练的GPR所决定的d-维度山脊线上。这一算法证实不管是低维还是高维组分空间,NEB算法确实沿着山脊线走向。同时这一算法也揭示了组分空间中GPR的潜在问题,即山脊线会在不希望的地方探测到。在某些情况下,这可能归因于GPR学习催化活性的方式。这个问题在一维山脊线上尤其明显。GPR学习到的非物理最大值影响一维脊的形状,使得GPR和脊检测的组合不适合在更高维度的组成空间中获得脊的可靠轨迹。
从使用NEB算法的结果中可看出该算法确实可以在组分空间中跟踪活性函数的最大值之间的脊。这些脊在更高维度上的确切形状和互连性仍未完全理解,且GPR由于在组成空间中受到超维单纯形的限制而显示出局限性。在更高维度组分空间中所有局部最大值是否通过山脊线连接也仍不能完全确定。五元组分空间中经典NEB算法似乎能够连接大多数最大值,这也表明,在更高维度空间中最大值仍然通过脊连接。
为了通过研究任意活性函数来进一步考察这种活性“路线图”的本质和模型的一般行为,作者选择选择了与“人工元素”相对应的动力学模型的随机生成参数来模拟不同的活性函数。尽管生成的人工元素的活性小得多,但活性“路线图”仍显示出山脊线,且使用ML NEB的元素替代策略可以在最大值之间进行探测。
使用NEB算法的缺点是找到的路径取决于初始和最终组成。为了规避这一点并进一步探索与生物进化路线图的相似性,作者提出了一种类似于定向进化(DE)的可能实验策略。图4显示了通过三元组分进行DE模拟的结果。DE以相对活性较高的Ir50Pt50开始,以显示如何进一步优化已表现出良好性能的催化剂。DE沿着山脊线前进并经过几个最大值。有趣的是,这条路径似乎被“困”在钯周围,沿着山脊线绕着钯转。所选的点不采样精确的最大值,也不太可能这样做,因为25 at%的相对较大且恒定的步长刚好适用于这些模拟。为了对路径上采样点之间的最大值进行采样,可以使用沿着路径的一维贝叶斯优化,或者仅仅是简单的直觉来预测最大值可能位于何处。在图4中,三元合成空间上的模拟仅显示了活性路线图的概念,但该策略在维度上是可扩展的。
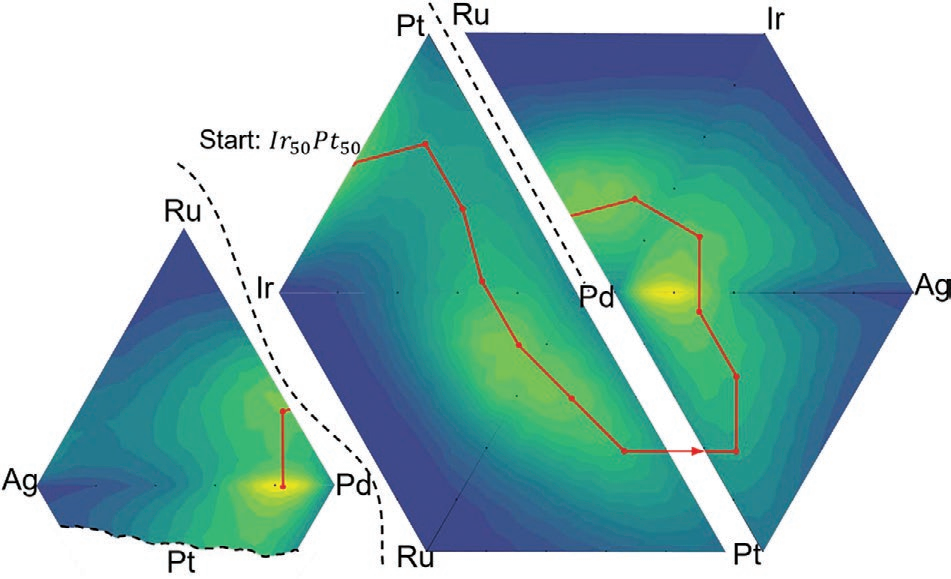
图4在Ir-Pd-Pt组成空间中从Ir50Pt50开始的DE模拟。
正如在这项工作中提出的,进化路线图只有一个拟合参数,即催化活性。影响路线图的其他拟合参数可能是稳定性或材料成本。为了解组成元素的稀缺性如何影响适应度,对Ag-Ir-Pd-Pt-Ru HEA组成空间的均匀5at%网格上的所有组成,将催化活性与稀缺性的度量(组成元素的生产速率与Pt生产速率归一化的倒数)进行绘制。结果如图5所示。帕累托最优是只能顾及提高催化活性或缓解元素稀缺性中的一种的主要原因。由于Ag的产量明显大于其余元素,因此帕累托前沿的大部分,即帕累托最优集合,基本上以不同比例的Ag-Pd二元合金形式出现。
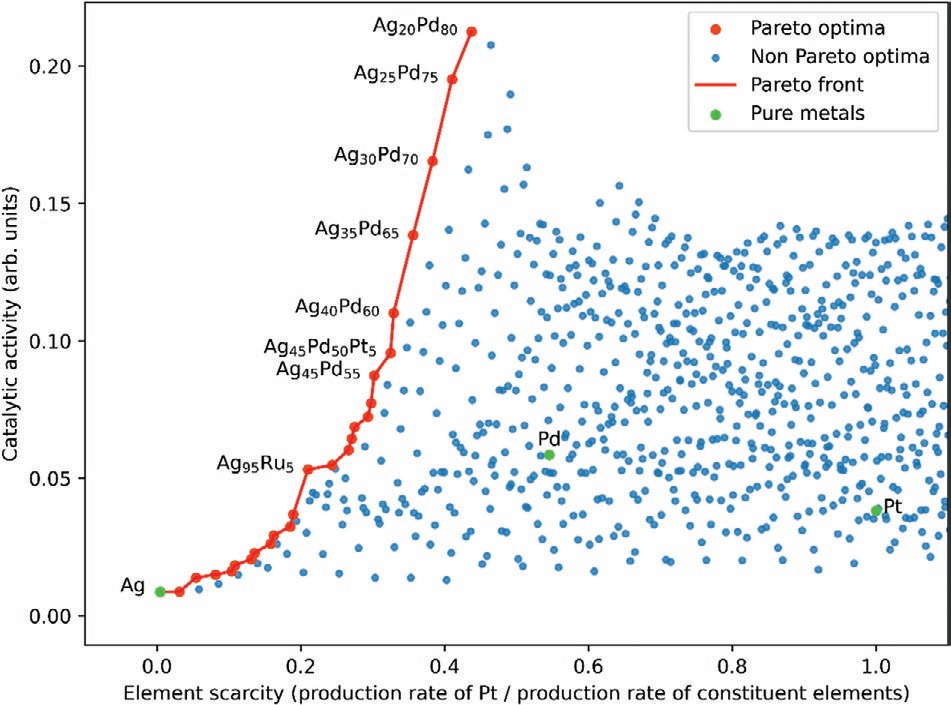
图5从Ag-Ir Pd-Pt-Ru的所有组成的5at%网格中找到的帕累托最优。
05、成果启示
该工作通过使用GPR方法和修正的NRB算法对HEAs组分空间进行模拟,并给出了催化活性最优者沿山脊线的分布,该分布也被山脊线检测算法证实。沿着催化活性的脊到组成空间的边缘,然后用一种元素替换另一种元素,并在三元组成上进行模拟的新策略,通过引入更多的适应度参数,适应度景观可以被改变以根据参数找到有效组合,不局限于只寻找最具催化活性的组合物。组合空间的进化行为可能会改变广阔空间的构思方式,并在寻找和设计新的催化剂时提供新的思维方式。
审核编辑:郭婷
-
能源
+关注
关注
3文章
2405浏览量
46134 -
机器学习
+关注
关注
67文章
8562浏览量
137208
原文标题:AEM:寻找最高催化活性的高熵合金组成
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
双原子催化剂突破固相转化反应绝缘屏障助力高能量锂电池

终结催化“中毒”烦恼,告别频繁更换:四方光电让NMP监测一劳永逸
华为携手产业伙伴斩获TM Forum卓越催化剂项目使命先锋奖
3552次循环突破!新型复合催化剂解锁锌电储能新纪元
钙钛矿太阳能电池的紫外光催化降解


瞬态吸收光谱技术从机理层面为光催化研究提供指导
瞬态吸收助力理解AQ(蒽醌)在H•−ORR光催化过程中的作用机制
LED 太阳光模拟器:光谱匹配AM1.5G 保障光催化活性测试的准确性

光谱匹配度对太阳能光催化反应器定量解析
太阳光模拟器 | 光催化材料测试的精准利器
瞬态吸收光谱技术揭示光催化过程关键机理,进而为g-CN基光催化材料的性能提升提供了新的策略
基于碳纳米材料的TPU导电长丝制备与性能研究

连续焦耳加热赋能三元协同催化剂,高效水处理方案来袭




评论