 原位表征揭示负载型金属间化合物Pd2Ga表面原子排布调控机制
原位表征揭示负载型金属间化合物Pd2Ga表面原子排布调控机制
全文速览
催化剂活性中心金属表面原子排布决定了反应物分子的吸附、活化及反应,因此对其精准调控是多相催化领域中的核心问题之一。本文利用反应金属-载体相互作用(reactive metal-support interaction,RMSI)原理对Pd2Ga金属间化合物纳米粒子表面结构进行调控,通过原位调节金属间化合物纳米粒子的还原程度可以实现由连续的Pd三原子(Pd3)向孤立的Pd单原子(Pd1)排布转变。
结合原位透射电子显微镜和原位谱学等原位表征手段,以及乙炔加氢反应揭示了Pd2Ga纳米粒子暴露面/表面Pd原子排布变化及其与反应性能关系,并进一步通过理论模拟揭示其结构转变机制。该工作有效地推进了RMSI驱动下金属间化合物表面结构转变的认知,为催化剂表面原子排布精准调控提供了新思路。
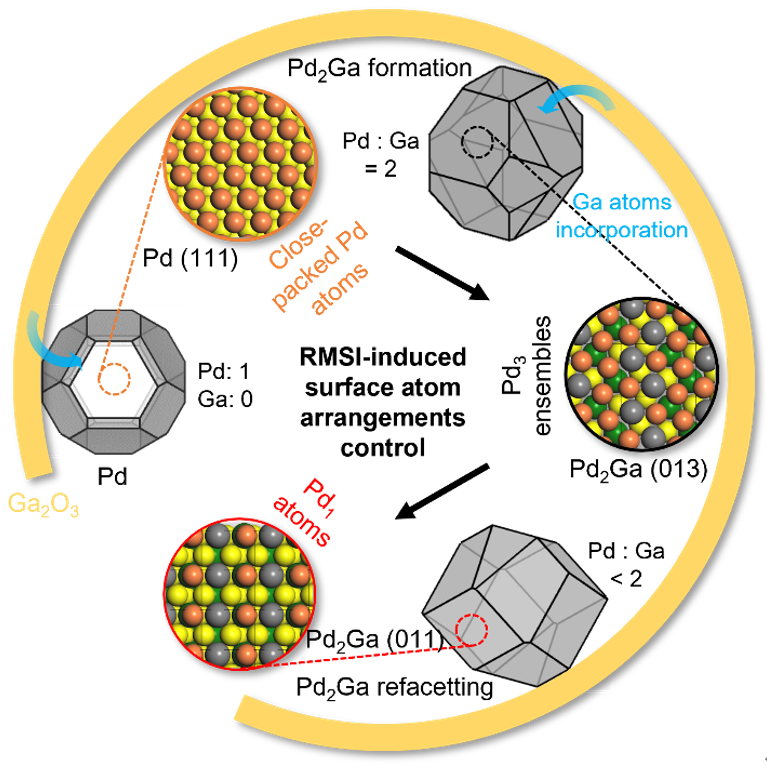
背景介绍
催化转化过程中,催化材料活性中心金属的几何结构决定了反应物的吸附构型/强度、中间产物在表面迁移/反应以及产物的脱附能力。在原子尺度调节活性金属表面几何构型可以实现催化反应的精准控制,进而获得优异的催化性能。前期大量的研究结果表明,通过调节活性金属组分和载体之间的强作用能力,惰性金属合金化以稀释活性金属在表面的分布、以及特定配位构型的金属配合物前体制备负载型催化剂等方式可以实现活性金属表面原子排布的调控。虽然这些活性原子组合结构已经表现出优异的性能,但是在不经过多步合成或使用复杂金属配合物前体的情况下,在原子尺度控制合成高度均匀且表面原子排布可控的催化剂仍存在巨大挑战。
金属间化合物因其原子高度有序的排布,理论上讲,在不同表面会表现出特异的活性金属原子排布结构,被广泛的应用到重要的催化反应中,如:选择性加氢、水煤气变换、一氧化碳氧化、烯烃环氧化和电催化氧还原反应等。PdGa纳米粒子作为一种典型的金属间化合物,分别表现出分隔的Pd原子三聚体和单原子Pd结构。
通过调控负载纳米粒子和载体之间的相互作用,实现金属间化合物纳米粒子具有特定的暴露面和表面原子排布,在理论上被认为是一种可行且简单有效的手段。但由于缺乏对金属间化合物形成和演变机制的理解,实现负载金属间化合物纳米粒子表面原子结构的精准调控仍存在挑战。
特别地,活性金属纳米粒子的表面结构和它们所处的化学环境密切相关。在真实的反应条件下和结构表征时催化材料所处的化学环境存在较大差异,这将会对活性金属结构的解析带来误导,从而不能建立准确的构效关系。因此在原位条件下,特别是耦合原位透射电子显微术和原位谱学方法等微观-宏观相结合的分析手段,研究催化剂结构的形成和演变是非常必要的。
研究目标
揭示反应金属-载体相互作用对负载Pd2Ga金属间化合物纳米粒子表面Pd原子排布调控机制及其与乙炔加氢反应性能关系。
图文精读

氧化镓负载钯催化剂在高温氢气气氛中可以通过RMSI生成Pd2Ga金属间化合物结构(Figure 1A)。首先通过水热法合成了棒状形貌的氧化镓纳米晶,在不同温度下焙烧获取不同的相结构,基于结构和形貌选择α-Ga2O3为载体制备负载型Pd基催化剂,接下来在不同温度氢气气氛中进行还原处理。
XRD结果表明还原后在100-200、300-500和600 oC分别得到了Pd、Pd2Ga和Pd2Ga/Pd5Ga3结构,这些结构可以通过位于46.7o的Pd(200)和44.2o/47.4o处的Pd2Ga(020)/(203)衍射峰识别(Figure 1B)。原位透射电镜结果显示升温还原过程中载体基本保持不变,Pd纳米粒子均匀分布在载体表面孔洞处,形成Pd2Ga后粒子尺寸略微增大(4.9±1.4 nm到5.8±1.5 nm,Figure 1D)。

如上所述,XRD确定了升温还原后Pd-Ga2O3催化剂的晶体结构演变,随后通过原位透射电镜(in-situ TEM)解析RMSI过程Pd纳米粒子微观结构变化。结果表明在200 oC氢气气氛(300 Pa)下还原得到了金属钯,纳米粒子以截角八面体形貌存在,主要暴露(111)、(200)及(110)晶面(Figure 2A);提高还原温度至400 oC,该纳米粒子转变为Pd2Ga结构,粒径略微增大并主要暴露(013)、(202)、(211)及(215)晶面(Figure 2D),原子模型结果展示了转变前后纳米粒子的形貌及暴露面变化(Figure 2C,F)。
傅里叶变换(FFTs)结果表明纳米粒子转变前后均为单晶结构,观察轴向由Pd [01]变为Pd2Ga [11](Figure 2B,E)。由于Pd-Ga合金在富钯区间存在多种金属间化合物结构且结构类似,其结构解析存在困难,因此通过ADF-STEM结合STEM模拟和原子尺度的能谱进一步精确解析生成的金属间化合物纳米粒子结构(Figure 2G-N)。
结果表明不同轴向的ADF-STEM结果与STEM模拟和原子模型完全对应,展示了由Pd和Ga原子序数差异造成的不同原子柱(Pd、Pd/Ga)在图像中的衬度和精细差异,如[13]轴向Pd原子柱的衬度明显高于临近Pd/Ga混合组成的原子柱,以及在[01]轴向不仅可以观察到不同原子柱的衬度差异,且可分辨原子柱在不同方向的衬度分布差异,总体呈现椭圆形并与STEM模拟和原子模型精确吻合。
能量色散X射线谱分析也在原子尺度上准确鉴别了Pd2Ga结构中Pd和Ga的分布(Figure 2N)。这些结果共同证明一定温度区间内RMSI调控负载型催化剂形成高度有序的Pd2Ga结构。
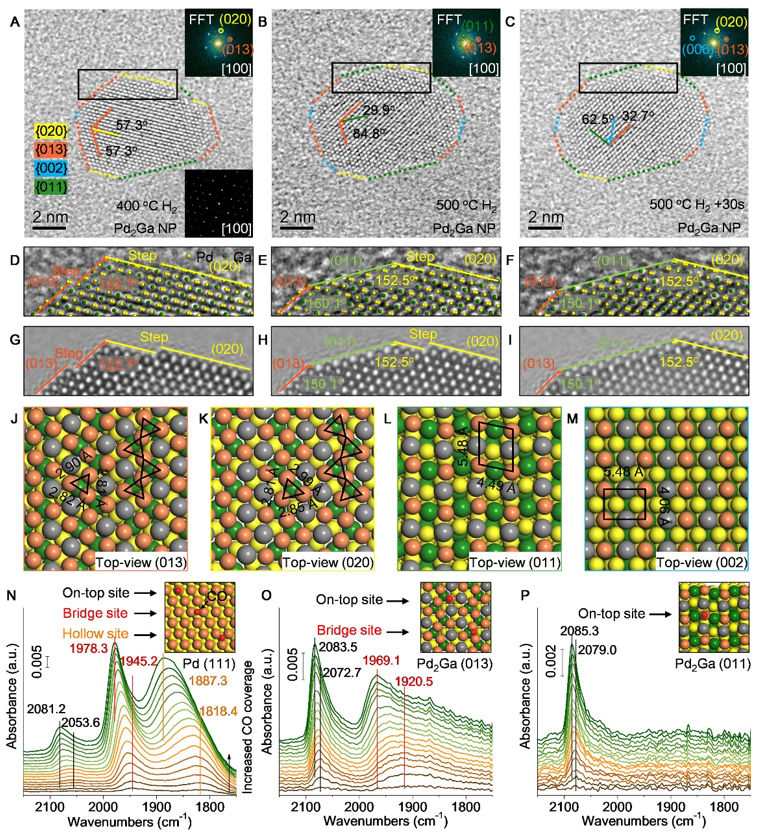
上述结果表明一定温度区间可以获得Pd2Ga纳米粒子结构,进一步通过环境透射电镜(ETEM)观察Pd2Ga纳米粒子初始形成及升温条件下的微观结构演变。Figure 3A-C展示了从[100]轴向观察的Pd2Ga纳米粒子在氢气气氛下400到500℃的变化,直观发现纳米粒子形貌在升温条件下发生了变化,通过电镜数据分析结合FFT以及高分辨模拟(Figure 3D-I)发现纳米粒子的暴露面从(013)和(020)转变为了(011)和(020),并且随着时间的延长更多的晶面发生了转变。
基于Pd2Ga的晶体结构分析纳米粒子形貌重构过程中的表面Pd原子排布演变(Figure 3J-M)。Pd2Ga(013)和(020)面是密排面且表面Pd原子排布类似,均为三个邻近Pd原子组成三角形排列(Pd3),然后通过顶点连接形成锯齿形。两种晶面上Pd原子之间的距离类似但存在略微差异,(013)面上相邻的Pd原子之间的距离为2.81、2.82及2.90 Å,而(020)面略微较大,为2.81、2.85及2.99 Å,均是线性排列的Pd原子之间距离最小,而锯齿形两端的Pd原子与中间的Pd距离较大。
Pd2Ga (011)和(002)晶面Pd原子的排布与(013)和(020)晶面完全不同,表现出分隔开的Pd原子结构(Pd1),最近的Pd原子距离分别为4.06、4.49及5.48 Å(Figure 3L-M)。因此,结果表明初始形成的Pd2Ga纳米粒子表面由连续的Pd3原子组成,升温条件下转变为Pd1。
我们利用原位TEM直接在原子尺度观察了Pd2Ga纳米粒子的微观结构演变,并解析其表面Pd原子排布变化,进一步结合谱学手段综合表征了负载Pd2Ga纳米粒子在升温条件下的表面结构演变。一氧化碳吸附漫反射红外(CO-DRIFTs)结果显示Pd2Ga生成后对CO的吸附发生了巨大的变化(Figure 3N-P),从开始的三种CO吸附共存(线式、桥式和空穴吸附)转变为只存在线式和桥式吸附。
空穴吸附峰的消失可能是Pd2Ga表面Pd原子间距离较大,Pd(111)晶面上邻近Pd原子距离为2.75 Å,而Pd2Ga(013)和(020)面上邻近Pd原子距离2.8至3.0 Å,因此CO可能优先桥式吸附在距离较近的两个Pd原子(2.8 Å)上,以及线式吸附在锯齿状末端的Pd原子(与邻近Pd原子距离为2.9~3.0 Å)上。
随后进一步提高还原温度至500 oC后,在Pd2Ga上只观察到了CO线性吸附峰,表明此时Pd2Ga表面只存在孤立的Pd1原子结构(Figure 3P)。可见,升温条件下Pd2Ga纳米粒子表面Pd原子排布的均匀转变是连续的Pd3转变为孤立的Pd1。

上述结果表明氢气气氛下负载的Pd2Ga纳米粒子在升温下发生表面重构现象,随后通过理论计算研究该现象发生的内在机制。理论计算得出Pd2Ga各晶面的表面能,其中密排面如(210)、(013)、(211)、(020)及(112)表现出较低的表面能(1.17到1.32 J m-2),而非密排面(223)、(230)、(002)及(011)等表面能较高(1.50到1.56 J m-2)。随后基于不同晶面的表面能构建了Pd2Ga纳米粒子的Wulff形貌(Figure 4B),其中近97%的表面被(210)、(013)及(020)等面占据。
我们根据前述原位透射电镜结果分别构建了两种由(210)、(013)、(020)、(202)和(400)、(002)、(011)面封装的Pd2Ga纳米粒子原子模型,探索不同大小(0.5至10.0 nm)Pd2Ga纳米粒子中Pd和Ga原子数量变化和差异。
如Figure 4C所示,(210)、(013)、(020)及(202)面封装的Pd2Ga纳米粒子随粒径增加Pd和Ga的原子比保持为2.0,与Pd2Ga结构原子比一致,而纳米粒子暴露(011)、(002)晶面时Pd/Ga比值均低于1.9,并且当粒子小于3纳米时该比值急剧减小(1.1-1.7),这可能是表面原子在总体原子中所占比例增加导致。
另外为了模拟RMSI过程中负载纳米粒子中Pd不变的情况,在控制Pd原子总数不变(3365)的同时改变Pd2Ga(~5 nm)纳米粒子的暴露面来研究Ga原子的变化。如Figure 4D所示,随着(011)和(002)面所占比例的增加,Ga原子的数量从1690增加到1897,Pd与Ga的比例也相应从1.99下降到1.78。
因此,这些结果表明通过RMSI形成的Pd2Ga纳米粒子初始主要暴露(210)和(013)等面,粒子中Pd和Ga的比例为2,随着还原温度的升高,更多的Ga原子被还原出来并进入到Pd2Ga纳米粒子中,导致表面重构和表面Pd原子排布变化。
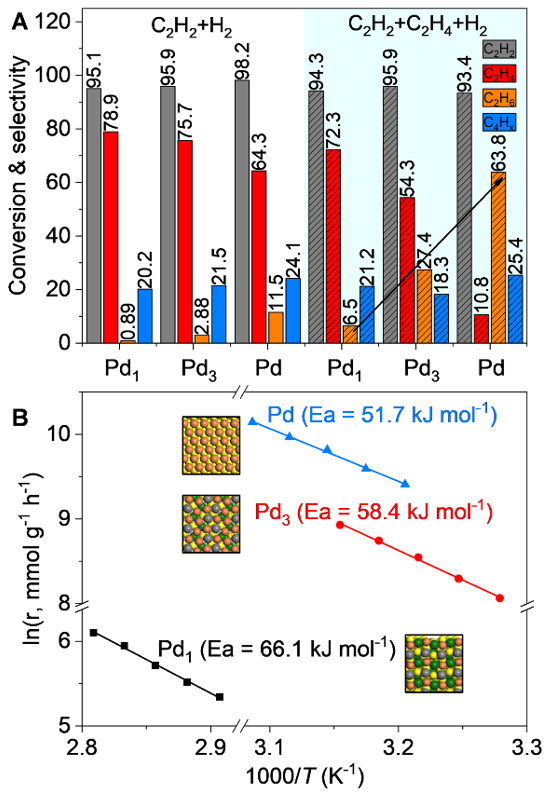
乙炔选择性加氢反应是结构敏感性反应,其活性和选择性会随Pd原子排布改变发生变化。因此,我们通过乙炔加氢反应来揭示三种表面Pd原子排布结构催化剂的性能差异。反应结果表明(Figure 5A)无乙烯的情况下,负载型Pd催化剂,即连续Pd原子排布结构在98.2%的乙炔转化率表现出11.5%的乙烷选择性,而Pd2Ga表面连续Pd3和Pd1结构分别表现出2.9%和小于1%的选择性。
在过量乙烯存在(20%)的情况下,连续Pd3和Pd位点的乙烷选择性分别从2.9%和11.5%增加到了27.4%和63.8%,而孤立的Pd1位点催化剂在94.3%的乙炔转化率下仍然具有很低的乙烷选择性(6.5%)和高的乙烯选择性。另外动力学实验结果表明随着表面Pd原子被逐渐分隔,乙炔加氢反应的活化能逐渐从51.7增大到58.4和66.1 kJ mol-1,催化剂的加氢活性下降。
心得与展望
通过原位透射电镜精准解析在程序升温还原条件下负载型Pd2Ga纳米粒子的结构及表面演变,并结合高分辨模拟和原位谱学等手段证明了在RMSI作用下纳米粒子表面Pd原子排布从连续的Pd3转变为分隔开的Pd1结构。
进一步基于理论计算揭示了该转变过程内在机制,较低温度初始形成的Pd2Ga纳米粒子由表面能较低的密排面封装且暴露连续的Pd3原子排布结构,随着还原温度的升高更多的Ga原子被还原并进入到纳米粒子中,由于温度和量不足以形成新相,这些原子倾向于排步在粒子表面以维持Pd2Ga金属间化合物的结构,从而导致纳米粒子的形貌重构和表面结构变化,在该情况下纳米粒子中的Ga原子含量高于化学计量比。
不同的表面Pd原子排布在乙炔加氢反应中表现出不同的性能,连续的Pd3原子排布表现出高的质量比活性,而Pd1结构具有更好的选择性和原子利用效率。该工作揭示了RMSI对负载金属间化合物表面结构的调控及其机制,为基于RMSI设计和精细调控负载型金属间化合物催化剂的表面原子排布提供关键的参考和依据。
审核编辑:刘清
-
傅里叶变换
+关注
关注
6文章
437浏览量
42562 -
XRD
+关注
关注
0文章
131浏览量
9056
原文标题:金属所张炳森团队Sci. Adv.:原位表征揭示负载型金属间化合物Pd2Ga表面原子排布调控机制
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
灯具挥发性有机化合物(VOC)鉴定
微小无铅钎焊接头中金锡化合物的形貌与分布:激光与热风重熔方法的比较
磷酸二氢锂的相对原子质量是多少
京东方华灿光电亮相2024功率及化合物半导体产业国际论坛

清纯半导体、中电化合物入选2024年度宁波市优质产品推荐目录!

格力电器将于6月投产全球第二座SiC全自动化化合物芯片工厂

SAC305-SiC复合焊料对金属间化合物的影响
西北工业大学研发出双层扭转金属硫族化合物层间角度可调
一种用于调控Ga2O3薄膜的表面电子结构的的热重组工程

PCB表面处理浸锡的优缺点有哪些
氮化镓是什么晶体,是离子晶体还是原子晶体
氮化镓是什么化合物类型
中电化合物荣获“中国第三代半导体外延十强企业”
苏州长光华芯化合物半导体光电子平台新建项目奠基仪式启动
清软微视周继乐:化合物半导体衬底和外延缺陷无损检测技术






 工商网监
工商网监
评论