 氟化打破氮化碳光催化全解水瓶颈
氟化打破氮化碳光催化全解水瓶颈

【研究背景】
使用颗粒光催化剂从水中生产氢气是一种实现大规模太阳能转换的低成本绿色技术。作为一种无金属的二维无机纳米材料,石墨碳氮化物(g-C3N4)通过加入小分子有机物作为空穴清除剂,表现出优异的水分解产氢能力。然而,长期以来,单相g-C3N4被认为不能进行光催化全水分解,因为g-C3N4的OER能力不足,无法直接从纯水中产生氧气。研究人员普遍将这种不足的OER能力归因于 g-C3N4上价带空穴的氧化能力弱。
通常,一步激发全水分解需要半导体的带隙大于 1.23 eV 的热力学要求,并且跨越HER和OER的氧化还原电位。然而,在能带隙大于2.0 eV且价带和导带位置完全满足水分解热力学要求的情况下,g-C3N4仍无法直接从纯水中提取O2,表明是未知因素而不是高价带位置阻碍了g-C3N4上的OER。对于g-C3N4,弄清楚阻碍OER的瓶颈以及如何绕过这样的瓶颈以实现可见光下高效的全水分解是至关重要的。
【成果简介】
江苏大学闫研、密歇根大学周鹏教授通过对C=O键的原位观察,确定了在g-C3N4上无法实现光催化全水分解的瓶颈。通过氟化,碳位被表面氟原子占据,中间C=O键大大减少,实现了O2的连续生成。
【研究亮点】
1.F-CN的制氢性能比本征g-C3N4的性能高一个数量级,
2.通过C-F相互作用优化了相邻N原子的析氧反应途径,有效避免了原始g-C3N4上过强的C-O相互作用或弱的N-O相互作用。
【图文导读】
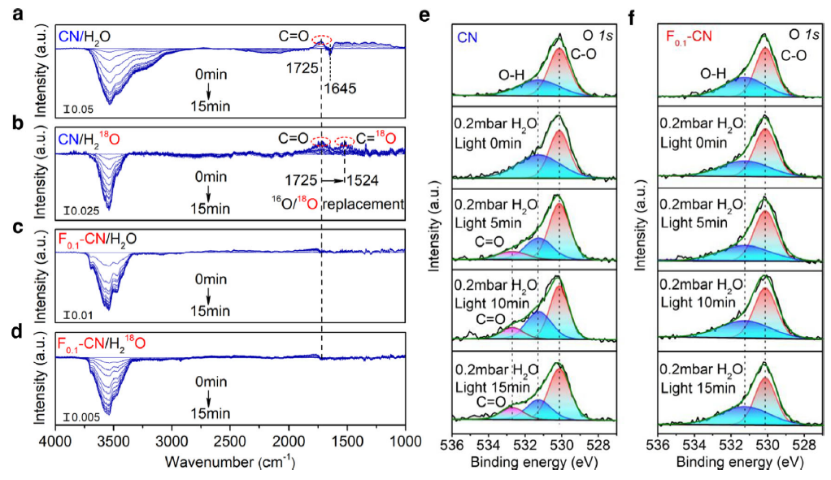
图1 a CN/H2O, b CN/H218O, c F0.1-CN/H2O和d F0.1-CN/ H218O的原位红外,e CN和F0.1-CN原位NAP-XPS O1s光谱。
如图1a所示,当使用CN/H2O样品原位照射时,从3700 cm-1到3000 cm-1的宽负吸收带出现并且强度随着照射时间的增加而增加。宽负带归因于O-H键的伸缩振动。相比之下,1645 cm-1处的弱负峰来自H2O分子的H-O-H的弯曲振动,代表在连续OER过程中表面-OH物种和H2O分子的损失。
O-H的伸缩振动信号远大于H2O分子的弯曲振动信号,表明发生在H2O/CN界面的OER主要以离解的OH形式存在。当CN替换为F0.1-CN或H2O替换为18O-标记的H218O时,观察到相同的特征(图1a−d)。更重要的是,随着照射时间的增加,在1725 cm-1处观察到归因于C=O伸缩振动的增加的正峰(图1a),表明 C=O 物种在 CN 表面上的形成。
当在其他相同条件下用18O标记的H218O 替换H2O时(图1b),1725 cm-1处的正峰和1524 cm-1处新生成的峰以16O/18O的形式出现,这证实了C=O的O源来自H2O。C=O的形成只能发生在CN上的碳位被氧化的情况下。用 F0.1-CN 替换CN时(图1c),在H2O/F0.1-CN 界面不再观察到正C=O信号(图1d),这有力地证实了CN的氟化可防止碳位点被氧化成C=O中间体。
NAP-XPS 也直接观察到通过氧化 CN 上的碳位点集体形成 C=O 态。在原始CN的 O1s光谱中,观察到位于 530.1 eV 和 531.3 eV 的两个主峰,分别对应于CO 和OH物种(图1e)。在白光照射下,观察到C=O在 532.7 eV 处出现,并且随着照射时间的增加强度增加(图1e)。
NAP-XPS 结果与原位 DRIFTS 观察结果一致(图1a、b),表明在OER过程中确实在H2O/CN界面形成了C=O中间态。F0.1-CN的O1s(图1f)光谱几乎没有变化,这进一步证明在F-CN上C=O的形成已大大减少。
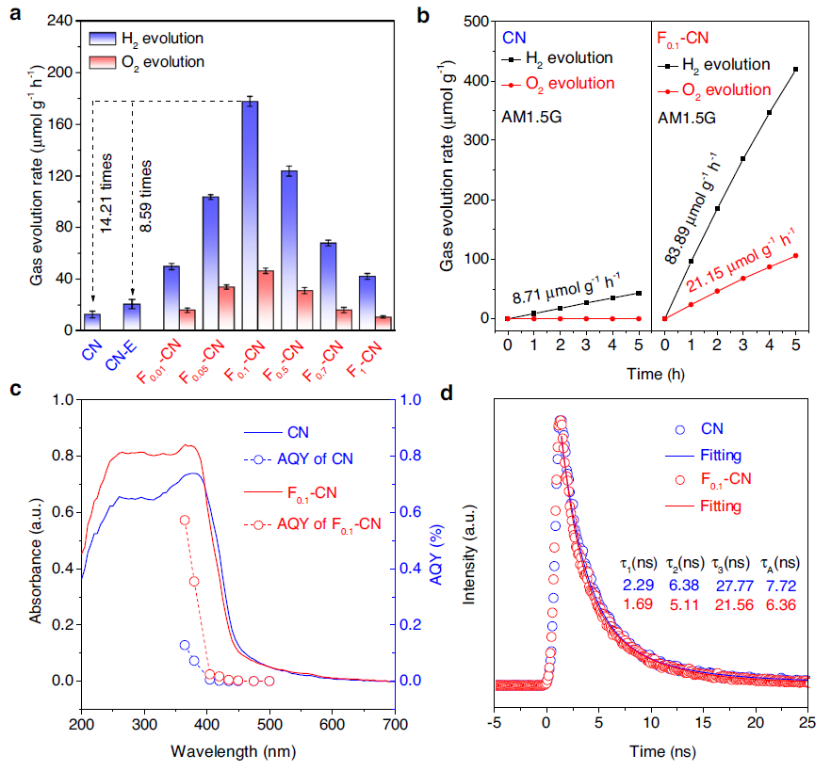
图2 a 光催化全解水性能,b 光催化生成H2和O2的时间曲线,c 原始CN和F0.1-CN的AQYs,d CN和F0.1-CN的瞬态荧光。
在白光(图2a)和 AM1.5 G 模拟太阳光照(图2b)下,在不含任何有机牺牲试剂的纯水中对 CN 和不同 F-CN 样品进行光催化全水分解实验。在白光照射下,原始CN催化剂仅表现出12.51 μmol g−1h−1的产H2性能,没有O2产生。水热处理后,CN剥离薄层样品(表示为 CN-E)仍未观察到O2产生。
在氟化处理后,所有F-CN催化剂在相同的实验条件下均表现出H2和O2释放能力,这随氟化程度而变化。F0.1-CN显示出177.79 μmol g−1h−1的产H2速率,分别比原始CN和CN-E高14.21倍和 8.59 倍,产O2速率为46.47 μmol g−1 h-1(图2a)。
在AM1.5模拟太阳照射下,F0.1-CN催化剂的H2产生速率为83.89 μmol g−1h−1,O2的产生速率为21.15 μmol g-1h-1(图2b)。图2c显示两个样品在365 nm处的 AQY为0.5718% (F0.1-CN)和0.1281% (CN)。与原始CN催化剂相比,F0.1-CN催化剂的荧光寿命没有显着延长(图2d)。寿命从7.72 ns减少到6.36 ns。
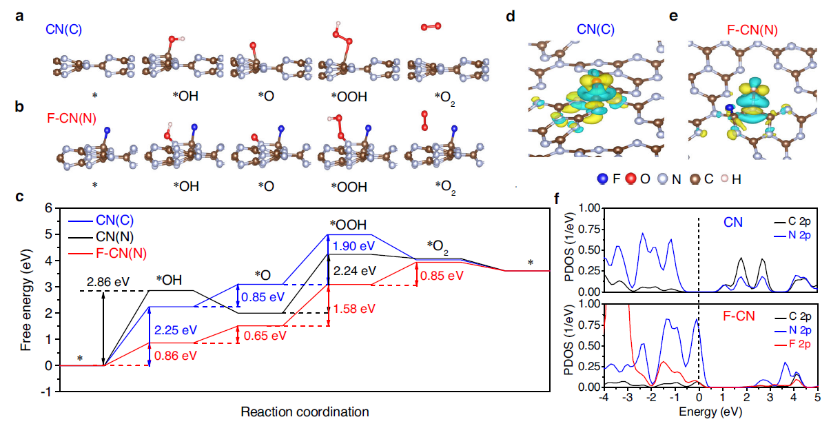
图3 a 在原始CN的C位点(记作CN(C))和 b 在F-CN的N位点(记作F-CN(N))上的水吸附和活化。c pH= 7和U = 0 V时CN和F-CN上OER的自由能。*OH中间体与催化剂表面的电荷密度差: d CN(C)和 e F-CN(N)。f CN和F-CN中C, N和F表面2p态的PDOS。
在原始CN中的C位点(图3a),N位点,F-CN中C-F键相邻的N原子作为反应位点(图3b)模拟水吸附和活化。计算的自由能分布表明,表面C位点上的 OER具有比CN中表面N位点(2.86 eV)更低的能垒(2.25 eV)(图3c ), 表明 C位点是原始CN中主要的OER反应位点。F原子占据C位后,C位饱和,对反应物或中间体呈惰性。
因此,相邻的双配位N位点是F-CN上唯一的催化中心。获得的反应途径表明,与原始CN中的表面C或N位点相比,F-CN中的表面N位点对 *→*OH (0.86 eV) 和*O → *OOH (1.58 eV) 的能垒低得多,表明F修饰确实可以提高F-CN上的 OER 活性(图3c)。
根据计算的电荷密度差异图,F-CN上 OER活性的提高归因于CN表面和*OH中间体之间更多的局部电荷分布,这显着促进了*OH 中间体的形成(图3e),并且也有效避免了原始CN中过强的C-O 相互作用(图3d)或弱的NO相互作用。
因此,F 修饰显着降低了决速步*OH的形成能。F修饰也显着促进了*OOH的形成(图3c)。F修饰使 N2p态向上移动到费米能级(图3f), 这可以归因于部分电子通过C位点转移到F位点。更正的 N 2p态保证N位点具有更高的氧化活性。
【总结与展望】
作者认识到表面碳原子被氧化为C=O是实现CN基催化剂全水分解的瓶颈。为了克服这个瓶颈,利用一种简单的表面氟化处理,通过形成C-F来抑制C=O积累,并通过激活相邻的N反应位点来降低OER势垒,从而使F-CN在可见光下具有全水分解的性能。
审核编辑:刘清
-
NAP
+关注
关注
0文章
2浏览量
7691 -
催化剂
+关注
关注
0文章
94浏览量
10830 -
XPS
+关注
关注
0文章
99浏览量
12566
原文标题:Nature子刊:氟化打破氮化碳光催化全解水瓶颈
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
香水瓶气密性检测仪怎么用-岳信仪器

盘点六氟化硫气体检测仪在电力全链条的布防要点

SN65LVDS94 LVDS 串行解串接收器:设计与应用全解析
30W氮化镓全电压认证方案

“芯”品发布|镓未来推出“9mΩ”车规级 GaN FET,打破功率氮化镓能效天花板!
钙钛矿太阳能电池的紫外光催化降解

聚光太阳光模拟器在光催化制氢中的应用

瞬态吸收光谱技术从机理层面为光催化研究提供指导
LED 太阳光模拟器:光谱匹配AM1.5G 保障光催化活性测试的准确性
热压烧结氮化硅陶瓷逆变器散热基板
光谱匹配度对太阳能光催化反应器定量解析
太阳光模拟器 | 光催化材料测试的精准利器
瞬态吸收光谱技术揭示光催化过程关键机理,进而为g-CN基光催化材料的性能提升提供了新的策略
全电压!PD 20W氮化镓电源方案认证款:U8722BAS+U7612B



评论