 阐明Pt单原子催化剂的轴向配体效应对碱性析氢反应的影响
阐明Pt单原子催化剂的轴向配体效应对碱性析氢反应的影响
研究背景
电催化析氢反应 (HER) 制备氢气是一种可有效解决碳排放问题的方案。研究者对酸性介质和碱性介质中的HER展开了广泛的研究。在强酸性介质中,为避免电极溶解,通常需要用贵金属基催化剂电催化质子还原。相比之下,多种催化剂在碱性电解质中表现出较好的稳定性。然而,碱性电解质中的HER动力学通常比酸性电解质中低几个数量级。此外,在碱性介质中,催化剂表面结构的敏感性也比在酸性介质中高得多。因此,促进碱性电解质中水还原的缓慢动力学对于降低绿色制氢的高过电位和相关能量损失至关重要。
开发高活性单原子催化剂 (SACs) 是应对上述挑战的一个有效的解决方案。揭示化学/环境-催化性能的关系对于设计高活性的SACs至关重要。目前,调控SACs活性中心周围化学环境的方法包括缺陷工程、退火、金属载体相互作用、引入团簇/纳米粒子等。虽然利用上述合成策略开发新材料取得了巨大进展,但苛刻的条件不可避免地打破了SACs活性中心的同质性,导致SACs活性中心的化学环境与催化性能之间的相关性的复杂性和不确定性。近年来,一些Pt-SACs对碱性HER表现出良好的活性,但通过可控修饰单原子Pt位点周围的化学环境来促进HER活性的探索尚属罕见。这促使研究人员设计Pt-SACs系统。在该系统中,Pt位点周围的化学环境可以被精确操纵,从而确定可靠的构效关系,并用于未来的催化剂设计。
成果介绍
新加坡国立大学Lei Wang和北京化工大学刘军枫(共同通讯作者)通过电沉积的方法将Pt单位点引入NiFe层状双氢氧根 (LDH) 纳米阵列上,发展了一种简单的辐照-浸渍方法来精确调整Pt-单位点上的轴向配体(如,−F,−Cl,−Br,−I,−OH),从而在保持Pt-SACs均匀性的基础上建立了良好的化学-环境/HER-活性关系。通过详细的光谱和电化学表征,表明Cl-Pt/LDH具有优越的HER性能:在1.0 M KOH中,达到10 mA cm-2的过电位为25.2 mV。过电位为100 mV时,质量活性高达30.6 A mgPt-1,分别是HO-Pt/LDH和商用20% Pt/C的5倍和133倍。在相同的条件下,HER的活性遵循Cl-Pt/LDH》F-Pt/LDH》HO-Pt/LDH》Br-Pt/LDH》I-Pt/LDH的顺序,证实了轴向配体对HER活性具有显著的影响。密度泛函理论 (DFT) 计算表明,由于第一电子亲和度高,Cl螯合Pt位点对OH*和H*都具有最佳的吸附亲和力,从而促进了缓慢的Volmer步骤 (水解离),这是碱性HER的典型动力学限制步骤。此外,以Cl-Pt/LDH和NiFe-LDH分别作为阴极和阳极催化剂,组装了基于膜电极组件 (MEA) 的水电解槽。在60°C下,达到1 A cm−2需要的电压为1.87 V,能量效率为80%。该研究证明了以原子精度设计SAC活性位点的重要性,为下一代催化剂的设计提供了理论支撑。相关工作以《Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction》为题发表在Nature Communications期刊。
图文介绍
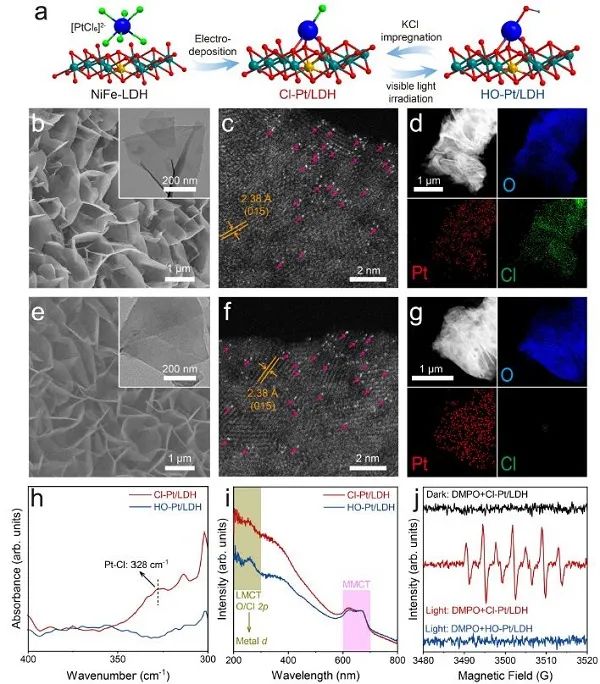
图1. Cl-Pt/LDH和HO-Pt/LDH的合成过程和结构表征。(a) 合成和配体交换过程的示意图;(b,e) SEM (插图:TEM);(c,f) HAADF-STEM,和(d, g) Cl-Pt/LDH和HO-Pt/LDH的元素映射;(h) Cl-Pt/LDH和HO-Pt/LDH的远红外光谱和(i) UV-vis DRS光谱;(j)光/暗条件下Cl-Pt/LDH和HO-Pt/LDH的EPR光谱。
调整Pt/LDH轴向配体的辐照浸渍过程如图1a所示。首先,通过水热法合成了NiFe-LDH纳米片阵列。然后,PtCl62-阴离子通过电沉积吸附到LDH表面,形成原子分散的Pt-位点。SEM和TEM图像显示,NiFe-LDH和Pt负载的LDH均呈现出横向尺寸约为500 nm、厚度约为10 nm的片状纳米阵列 (图1b,e)。通过像差校正的高角度环形暗场扫描透射电镜(AC-HAADF-STEM,图1c)未检测到亚纳米簇或纳米颗粒,证实了Pt在Cl-Pt/LDH中以原子级分散。此外,Pt和O在纳米板上的均匀分布表明Pt原子均匀地分散在LDH衬底上 (图1d,g)。同时,与NiFe-LDH的 (015 )面相对应的晶格条纹的面间距约为2.38 Å,与XRD结果一致。为了交换Cl配体,用白光照射Cl-Pt/LDH (3.75 mW cm−2;30分钟)。照射后Pt原子的原子分散没有变化 (图1f),但Cl信号的强度大部分减弱,表明Cl配体被成功去除。此外,辐照后远红外光谱中329 cm−1处的信号减少进一步证实了Pt-Cl键的丢失 (图1h)。初步将Cl的损失归结为OH- 作用下的阴离子交换,得到HO-Pt/LDH的产物,随后用X射线吸附光谱法确定其结构。在Cl-Pt/LDH和HO-Pt/LDH的紫外-可见漫反射光谱 (UV -vis DRS) 中,也观察到配体-金属电荷转移 (LMCT) 诱导的吸收带(~200 ~ 300 nm)发生了明显的变化 (图1i),而金属-金属电荷转移 (MMCT) 没有变化,说明辐照后只发生了配体交换。如图1j所示,当Cl-Pt/LDH与自由基清除剂5,5-二甲基-1-吡罗啉N-氧化物(DMPO)自发暴露时,会产生自由基信号,这可能是由于Cl配体通过光生空穴氧化成Cl自由基,然后被DMPO捕获形成DMPO+●自由基。相反,在黑暗中观察到Cl-Pt/LDH样品没有自由基信号,而在光照下观察到HO-Pt/LDH样品符合预期。
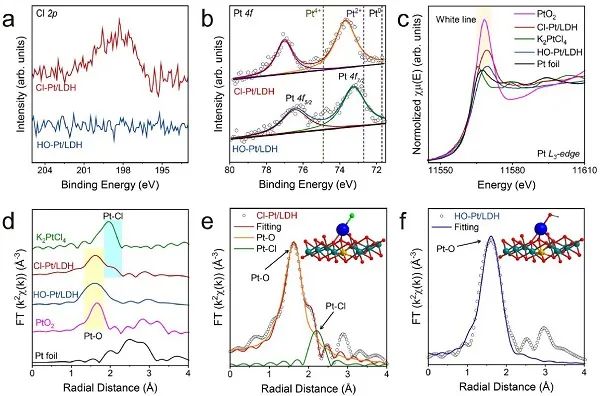
图2.电子态和坐标结构的表征。(a) Cl 2p和 (b) Pt 4f的高分辨率XPS谱;Cl-Pt/LDH, HO-Pt/LDH, Pt箔,K2PtCl4和PtO2在Pt L3-边的XANES光谱; (d) 对应的FT-EXAFS光谱; (e) Cl-Pt/LDH和(f) HO-Pt/LDH的EXAFS拟合曲线。
采用XPS分析了Cl-Pt/LDH和HO-Pt/LDH的化学组成。光照后,Cl 2p信号消失为完全的配体交换提供了进一步的证据 (图2a)。如图2b所示,Cl-Pt/LDH和HO-Pt/LDH中Pt-原子的4f7/2峰都位于Pt2+ (72.7 eV) 和Pt4+ (74.9 eV)之间,说明Pt-原子的平均价态在+2和+4之间。Cl-Pt/LDH中Pt 4f电子的结合能略高于HO-Pt/LDH中的结合能,这与先前观察到电子从Pt转移到Cl配体的结果吻合。用X射线吸收精细结构 (XAFS) 光谱研究了Cl-Pt/LDH和HO-Pt/LDH中单原子Pt位点的配位环境和电子结构。如图2c所示,Cl-Pt/LDH和HO-Pt/LDH的白线都位于K2PtCl4 (Pt2+) 和PtO2 (Pt4+) 的白线之间,说明Cl-Pt/LDH和HO-Pt/LDH中Pt原子的价态在+2 ~ +4之间,与上述XPS结果一致。Pt在Cl-Pt/LDH和HO-Pt/LDH中的价态分别为2.78和2.08,说明Pt-SACs的电子结构存在强配体效应。利用傅里叶变换(FT) k2加权扩展X射线吸收精细结构 (EXAFS) 光谱进一步分析了Pt-SACs的配位环境。如图2d所示,在~2.50 Å处,Cl-Pt/LDH和HO-Pt/LDH均未检测到Pt-Pt相互作用,说明Pt原子在这些样品中呈单原子分散状态。此外,Cl-Pt/LDH在1.60 Å和2.06 Å附近有两个明显的峰,分别与PtO2的Pt-O路径和K2PtCl4的Pt-Cl路径有关。相比之下,HO-Pt/LDH中仅在~1.60 Å处有一个Pt-O样峰,说明光照后Cl配体被完全取代。
Cl-Pt/LDH的最佳拟合结果表明,FT-EXAFS谱中位于Pt l3-边缘的1.60Å处的主峰可以归因于Pt-O第一配位,而在2.06 Å处的次峰可归因于Pt-Cl第一配位 (图2e)。根据Pt l3-边的Cl-Pt/LDH的EXAFS拟合参数,第一个配位球内的O原子位于2.02 Å,配位数为3.02,表明Pt位为四面体。此外,在2.29 Å处的Cl原子估计配位数为0.93,被认为是Pt上的垂直配体。综合考虑所有因素,提出了Cl-Pt/LDH最可能的结构,如图2e内嵌所示。对HO-Pt/LDH的类似分析表明,它的第一配位球中只有O原子,总配位数为3.60,证实Cl配体被OH-取代 (图2f),Pt位没有发生其他变化。
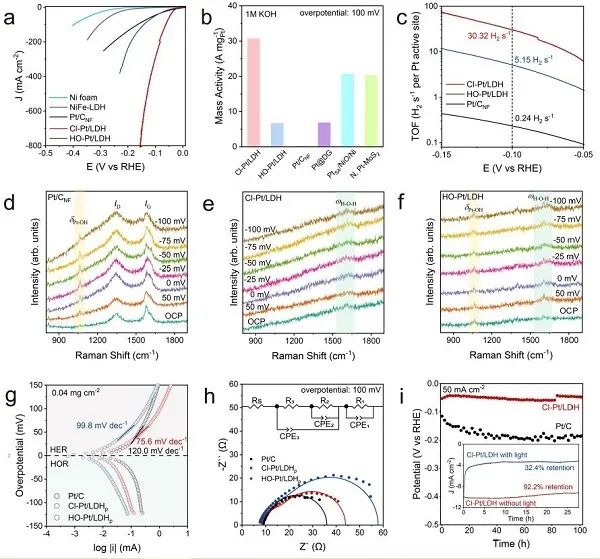
图3. 碱性介质中的HER性能。(a) 泡沫镍、NiFe-LDH、Pt/C、Cl-Pt/LDH和HO-Pt/LDH的HER极化曲线;(b) 本研究中Pt基催化剂的活性,以及其他地方报道的最先进的SACs: Pt@DG, PtSA/NiO/Ni和N, Pt-MoS2;(c) Pt基催化剂的TOFs图;(d) Pt/CNF,(e) Cl-Pt/LDH和(f) HO-Pt/LDH的Operando拉曼光谱;(g) Pt/C, Cl-Pt/LDHp和HO-Pt/LDHp的微极化曲线;(h) Pt基催化剂的EIS Nyquist图;(i) 阴极电流密度恒定为50mA cm−2时,Cl-Pt/LDH和商用Pt/C的电位-时间曲线。
如图3a所示,在所有测试的Pt-SACs中,Cl-Pt/LDH表现出优越的HER活性和~100%的H2法拉第效率,达到10 mA cm−2、100 mA cm−2和200 mA cm−2的电流密度的过电位分别为25.2 mV、51.9 mV和72.3mV,优于Pt/CNF (27.4 mV、164.9 mV和252.0 mV)和HO-Pt/LDH (41.5 mV、142.5mV和189.5 mV)。在100 mV过电位下,归一化Pt负载时Cl-Pt/LDH的质量活性估计为30.6 A mgPt−1(图3b),显著高于HO-Pt/LDH (6.6 A mgPt−1)、Pt/CNF (0.2 A mgPt−1)和其他论文报道的最先进的Pt-SACs。此外,在过电位为100 mV时,Cl-Pt/LDH (30.3 H2 s−1) 上Pt位点的翻转频率 (TOFs) 分别是HO-Pt/LDH (5.1 H2 s−1) 和商用Pt/CNF (0.2 H2 s−1)的5.9倍和126倍 (图3c)。
采用原位拉曼光谱法研究了氧化还原过程中表面物种及其化学键的变化。如图3d所示,在Pt/CNF上,HER仅在1062 cm−1处出现Pt-OH峰。因此,在催化条件下,*OH的解吸是缓慢的,这与之前的观察结果一致。相反,在相同条件下,Cl-Pt/LDH上没有观察到这种明显的Pt-OH峰 (图3e)。在~1630 cm−1处只有细微的峰值,这可能是由于吸附水的H−O−H弯曲模式,表明Volmer步骤和随后的*OH脱附步骤加快,导致HER动力学增强。HO-Pt/LDH中也存在吸附水的细微峰值,这与HO-Pt/LDH较Pt/C活性增强相一致。注意,虽然HO-Pt/LDH上的HER中也发现了少量的Pt-OH峰(图3f),但其强度不随过电位的变化而变化,这表明−OH是轴向配体,而不是来自水还原。总之,Pt-SACs上HER活性的提高可能源于Volmer步骤的加速。在此条件下,Cl-Pt/LDHp的Tafel斜率 (76.5 mv dec−1) 明显低于HO-Pt/LDHp (99.8 mV dec−1) 和Pt/C (120.0 mV dec−1),表明Cl-Pt/LDHp的迟缓Volmer步骤明显增强,RDS可能是Volmer步骤和Heyrovsky步骤的混合 (图3g)。在相同条件下,在电流密度为50 mA cm−2 和500 mA cm−2时,对Cl-Pt/LDH和Pt/C进行了稳定性测试 (图3i)。在100小时的测试期间,Cl-Pt/LDH比商用Pt/C催化剂表现出了更好的耐久性,证明了Pt-SACs在碱性HER条件下化学结构稳定。
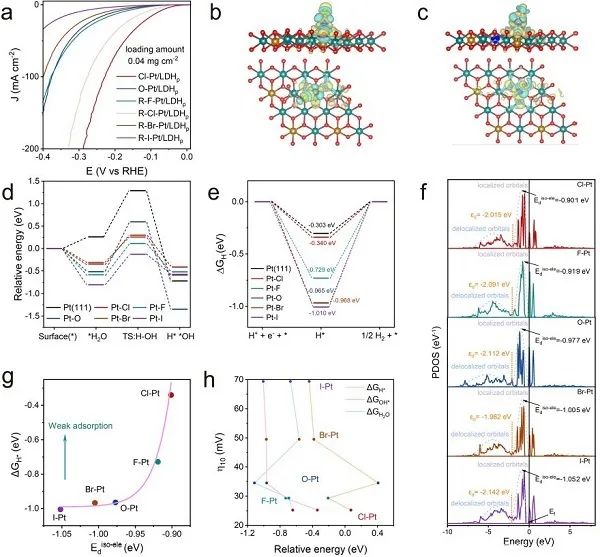
图4. 轴向配体效应及理论研究。(a) 不同轴向配体Pt-SACs的HER极化曲线;(b) Cl-Pt/LDH和(c) HO-Pt/LDH的计算模型和局域电场分布;(d) 计算水解离动力学的能垒和 (e) H*在Pt-SACs表面的吸附自由能;(f) 计算Pt-SACs的Pt 5d能带结构;(g) Eiso-ele d和ΔGH*的关系;(h) 碱性HER过电位 (在10mA cm−2) 与变量的关系。
不同轴向配体Pt-SACs的碱性HER活性随卤素原子的第一电子亲和度而变化 (图4a)。据EXAFS拟合结果建立了X-Pt/LDH (X = -F,-Cl,-Br,-I,-OH) 的计算模型。如图4b,c所示,在Pt-X键合区域观察到明显的电荷重分布,这可能是HER活性增强的原因。对碱性HER在Pt-SACs上的逐级反应能垒进行了模拟,包括水解离的Volmer步骤、*OH的解吸以及随后的H*转化为H2。对于Volmer步骤 (图4d),所有卤素配位Pt/LDHs都比Pt(111)具有更强的水吸附能力和更大的水分解焓,导致Volmer步骤加速。其中Cl-Pt/LDH在Volmer步的能垒最小 (0.073 eV)。另一方面,Cl-Pt-位点在Pt-SACs中也表现出最优的H结合能,导致H*转化为H2的动力学最佳 (图4e)。注意,虽然Pt(111) 的H结合能比Cl-Pt/LDH略优化,但它的水解离步骤可能较慢,导致整体HER动力学较慢。综合结果表明,H结合能并不是碱性HER的唯一描述符,OH结合能也起着重要作用。随后,计算了孤立电子(Eiso-ele d)占据的窄Pt-5d轨道的平均能级,分别为- 0.901,- 0.919,- 0.977,- 1.005和- 1.052 eV (图4f)。这与上述XANES的结果一致,在Cl-Pt/LDH中,由于Cl配体的第一电子亲和度最大,Pt的价态最高。根据d波段理论,表面中间体的吸附性能与催化剂的电子结构直接相关。配体在Pt单原子上的第一电子亲和度的提高可能会增加Eiso-ele d,从而进一步减弱对氢的吸附。Eiso-ele d最高的Cl-Pt与H*中间体的相互作用最弱,因此H*更容易解吸附并形成H2 (图4g)。总的来说,计算结果与实验观察一致,用轴向配体修饰Pt单位点的电子结构是调节HER活性的有效策略。综上所述,X-Pt/SACs的碱性HER活性随着配体第一电子亲和度的增加而增加,因为*H和*OH的吸附能都是碱性介质中HER活性的主要描述符 (图4h)。
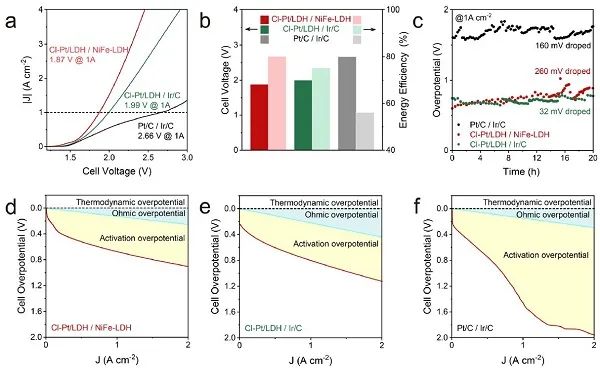
图5. 在MEA电解槽中电催化HER。(a) 分别以Cl-Pt/LDH / NiFe-LDH、Cl-Pt/LDH / Ir/C和商用Pt/C/Ir/C为阴极和阳极的MEA电解槽的LSV曲线;(b) 图5a中电流密度为1 A cm−2时的电压和能效;(c) 在60 °C下,电流密度为1 A cm−2时,MEA电解槽电解水的稳定性试验;(d) Cl-Pt/LDH/NiFe-LDH;(e) Cl-Pt/LDH/Ir/C;(f) 商用Pt/C/ Ir/C的欧姆过电位和活化过电位;Cl-Pt/LDH和Pt/C的Pt负载分别为1.01 × 10−2和0.8 mgPt cm−2。
为了评估Cl-Pt/LDH作为阴极催化剂在实际操作条件下的适用性,以NiFe-LDH为阳极,构建了基于阴离子交换膜的膜电极组件 (MEA) 电解槽。建立了以Pt/C和Ir/C为基础的MEA电解槽,并进行了对比试验。在60°C下获得了电流-电压曲线 (图5a)。在没有进一步优化的情况下,基于Cl-Pt/LDH / NiFe-LDH的电解槽在电流密度为1.0 A cm−2 时的槽电压 (1.87 V ) 比Cl-Pt/LDH / Ir/C (1.99 V) 和Pt/C / Ir/C (2.66 V) 的电解槽低得多。在此条件下,它们对应的能量转换效率分别为80%、75%和56% (图5b)。如图5c所示,Cl-Pt/LDH/Ir/C电解槽具有良好的稳定性。从图5d-f可以明显看出,Cl-Pt/LDH/NiFe-LDH或Cl-Pt/LDH/Ir/C的活化过电位远低于Pt/C / Ir/C,这是由于Cl-Pt/LDH具有较好的HER活性,从而提高了能量效率。
总结
作者将Pt-SACs锚定在具有不同轴向配体 (−F,−Cl,−Br,−I,−OH) 的NiFe-LDH纳米阵列上,通过简单的辐照浸渍过程制备了高活性碱性HER电催化剂。Cl-Pt/LDH在Pt- SACs和商用Pt/C中表现出最高的HER活性。Cl-Pt/LDH对碱性HER活性的增强是由于轴向配体的第一电子亲和度最大,对Pt单位点产生了强烈的电子效应。DFT计算表明,在单原子Pt位点上引入轴向配体可以调节Pt的d轨道孤对电子的平均能级,进而调节H*和*OH的吸附能。最后,在基于MEA的碱性水电解槽中评价了Cl-Pt/LDH的反应速率,获得了高达80%的能源效率,证明了其在制氢方面的应用前景。该工作凸显了调控催化中心周围化学环境的优势,其合成策略和轴向配体效应都可以用于指导未来SACs的设计,提高大规模绿色制氢的性能。
审核编辑 :李倩
-
电极
+关注
关注
5文章
806浏览量
27158 -
电解质
+关注
关注
6文章
803浏览量
20014 -
催化剂
+关注
关注
0文章
92浏览量
10284
原文标题:Nature子刊:阐明Pt单原子催化剂的轴向配体效应对碱性析氢反应的影响
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐





 工商网监
工商网监
评论