 通过溶剂化作用调控电解液性质和氧化还原反应
通过溶剂化作用调控电解液性质和氧化还原反应
研究背景
任何电化学装置的核心都是导离子电隔电子的电解液,其离子电导率和转移数决定了装置的功率上限。由于电解液与两个电极都接触,因此它还必须对电极材料和工作电压表现出高度的(电)化学稳定性,以防止降解反应。电解液的溶剂化特性在决定转化化学反应途径和动力学方面也起着至关重要的作用,例如 Li–S和 Li–O2电池。鉴于其在核心作用,电解液设计将对下一代电化学设备的开发至关重要,电池的下一个重大进步可能是受到新电解液发现的推动。例如,无论是电解液还是固态电解液的突破,这种突破都可能会促进锂金属电池产业化。
为了加快电池性能的提升,需要加深对电解液中分子过程的基本理解,以实现合理的设计。鉴于SEI由电解液的分解产物组成,哪些电解液成分可以为锂金属负极提供稳定的 SEI?此外,富镍正极材料很容易分解电解液,作者如何设计电解液以使其对高反应性富镍、高压正极稳定?此外,除了 Li-S 和 Li-O2等锂离子电池在充电和放电过程中会发生显着的相变;作者如何设计贫电解液环境和电池结构,使这些相变能够在高活性材料负载下发生?最后,传统液体电解液的离子电导率和低转移数限制了锂离子电池的最大功率;作者如何提高离子电导率和转移数以实现超快充电和高功率设备?
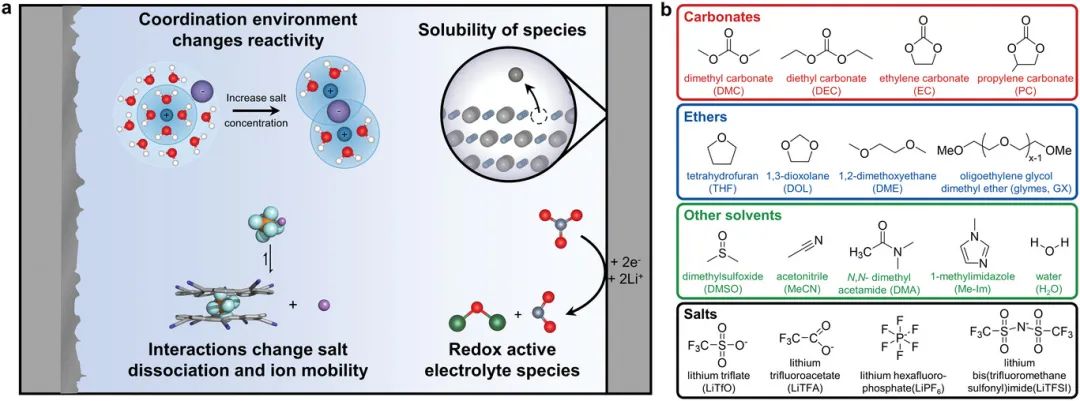
图1 a) 电解液中分子间相互作用可能对电池性能产生的关键影响概述。b) 文中将考虑的典型溶剂和盐的化学结构概述。
成果简介
在这篇mini综述中,作者强调可以调控常见的液态电解液(如碳酸盐和醚基)中的分子间相互作用(图1a)来显著提高电池性能的(图1b)。这种理解,再加上新材料的发现,对于下一代电解液能够提高电池性能至关重要。在第2节中,作者考虑了反应物、产物和反应中间体的溶解度如何影响电池反应途径和动力学。在第3节中,作者展示了电解液物种的配位环境对其(电化学)稳定性和反应性的深刻影响。最后,在第4节中,作者考虑了电解液内的分子间相互作用如何改变离子电导率以及离子迁移机制。
研究亮点
阐明了溶解自由能与溶剂化自由能的关系
举例说明了溶解度对Li–O2和Li–S系统性能的影响
利用不同手段改变溶剂化相互作用调控电解液中物种的反应性。
图文导读
Li+溶剂化基本概念
首先,作者提供了一些关于离子溶剂化热力学的理论背景和用于评估电解液中关键分子间相互作用的实验技术。Li+离子在有机电解液中的溶剂化主要是由溶剂上的给电子基团与 Li+离子之间的强焓相互作用驱动的。Li+的溶剂化吉布斯自由能() 定义为溶剂化 Li+和真空中孤立的 Li+之间吉布斯自由能的差异(图 2a)。由于真空中孤立的Li+的能量是恒定的,因此受 Li+、溶剂以及电解液中其他离子之间的焓和熵相互作用的调控。在有机电解液中,通常为 ≈−5至−5.6 eV,而是≈−0.5 至 −1 meV/K (−T在 25 °C时约为 +0.15 至 +0.3 eV),因此 Li+离子的溶剂化是由溶剂上的给电子基团(如碳酸盐和醚)与 Li+之间的强焓相互作用驱动的。
这种给电子相互作用可以通过 Gutmann 供体数 ( DN )、等溶剂化参数得到很好的描述,这些参数为纯溶剂和阴离子提供了易于比较的值,以衡量溶剂-阳离子和阴离子-阳离子相互作用的强度。另一方面,Li+的溶剂化在有机溶剂中,将溶剂分子构造成紧密结合的溶剂化壳,这将 Li+溶剂化的熵限制为 ≈-0.5 至-1 meV/K。例如,二甲基亚砜 (DMSO) 形成一个紧密的第一溶剂化鞘,每个 Li+周围有≈4 个 DMSO 分子。
假设每个 DMSO 分子与Li+配位限制的构型熵与冷冻的 DMSO 相当,作者可以估计由 DMSO 配位Li+引起的熵减少为4 × ΔSfus =−2.0 meV/K(其中ΔSfus=−0.5 meV/K),这可以很容易地解释测量的在DMSO 中为 -0.61 meV/K。除了溶剂化鞘中给电子焓和结构熵的影响之外,还有许多因素影响,例如电解液中Li+和阴离子之间的库仑相互作用和电子供体相互作用以及第二层溶剂化鞘。电解液中存在的分子间相互作用的多样性,使调控Li+的溶剂化能成为了可能。
要注的是意,锂盐在电解液中的溶解度不受溶剂化吉布斯自由能()的调控,而是由溶解吉布斯自由能()决定,其中包括锂盐晶格能这一附加项(图2a)。例如,LiTFA可以在乙腈 (ACN)中溶解度>2 m,而 LiNO3的溶解度≈0.3 m,尽管TFA− (≈34 kcal/mol)的DN值高于 NO3− (DN = 21 kcal/mol),这是因为LiTFA的晶格焓比LiNO3低(777 kJ/mol vs 823 kJ/mol)。
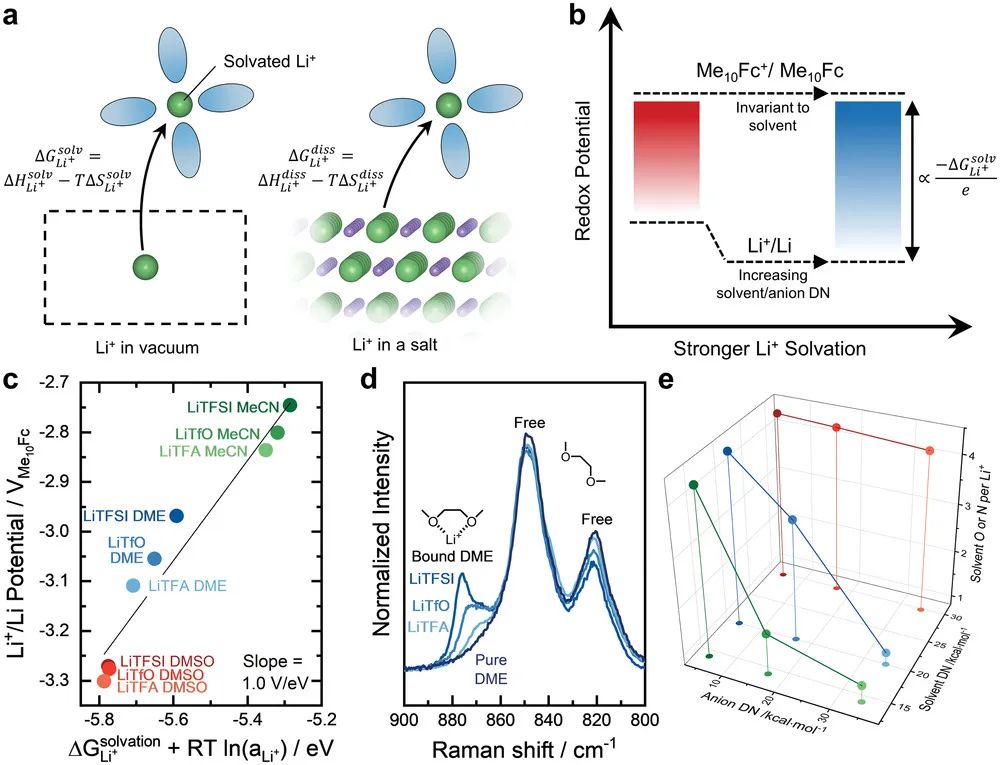
图2. a) 描述Li+溶剂化吉布斯自由能和溶解吉布斯自由能定义的示意图. b) 描述 Li+/Li 和 Me10Fc+/Me10Fc 氧化还原电位之间与溶剂化吉布斯自由能的差异,其中由于更高的溶剂或阴离子 DN而导致的更强的Li+溶剂化作用将 Li+ /Li 电位转移到更负的值。c) 将Li+/Li vs Me10Fc 的氧化还原电势与DFT计算的Li+溶剂化强度作图,该强度由离子电导率实验的Li+活性校正。d) DME中0.5 m LiTFSI、LiOTf 和 LiTFA 混合物的归一化拉曼光谱。e) 0.5 m LiTFSI (DN = 5.4 kcal/mol)、LiOTf (DN = 16.9 kcal/mol) 和 LiTFA (DN ≈ 34)与 MeCN (DN = 14.1 kcal/mol)、DME (DN = 20 kcal/mol) 和 DMSO (DN = 29.8 kcal/mol) 的混合物中Li+配位的溶剂中氧或氮原子的数量)).
Li+溶剂化的热力学参数可以通过量热法和电化学方法进行实验测定。锂盐(LiX)的溶解焓可以使用量热法测量,该量热法可以与盐的溶解吉布斯自由能相结合测定溶解熵。为了计算单离子溶解热力学(例如),必须做出额外的假设,其中最常见的是,假设四苯基胂(Ph4As+)和四苯基硼化物(BPh4−)具有相同的溶解吉布斯自由能、焓和熵。这种假设依赖于庞大的酚基团来有效地屏蔽带电离子中心对周围溶剂的影响,并且通常被认为是合理的,尽管据报道Ph4As+和BPh4−的溶剂化差异高达≈8%。基于溶解盐的晶格形成能的热力学路线,这些单离子溶解吉布斯自由能、焓和熵可以进一步与单离子溶剂化吉布斯自由能,焓和熵相关。
更实际地,依赖于电解液的氧化还原电位,如Li+/Li和十甲基二茂铁(Me10Fc),以确定溶剂化的单离子吉布斯自由能的趋势。在这种情况下,假设Me10Fc的溶剂化能不会随着其氧化还原状态而改变(由于其结构将氧化还原中心与电解液屏蔽),其电离能也不会随着溶剂化而改变,从而使其在所有溶剂中的氧化还原电势在绝对能级上保持恒定。根据这一假设,含有感兴趣离子的氧化还原对和Me10Fc的氧化还原偶之间的差异与离子的溶剂化吉布斯自由能成比例(图2b)。例如,Li+/Li和Me10Fc+/Me10Fc氧化还原电位之间的差异()与溶剂化吉布斯自由能有关。
其中e是基本电荷,C是由与电解液无关的形成能量项给出的常数,其中。因此与Li+溶剂化能的变化线性相关。最近,密度泛函理论(DFT)等计算技术提供了单离子溶剂化能的计算方法,其中Li+溶剂化环境包括明确定义的第一溶剂化壳层,然后将其嵌入隐式溶剂模型中。
值得注意的是,将DFT计算的Li+溶剂化能与实验测量的Li+/Li氧化还原电位进行比较,产生了斜率为1.0 V/eV的强线性相关性(图2c),表明这两种技术之间的定量一致。作者进一步发现,相应阴离子DN的增加可能导致在相同的溶剂中更负的,这可以通过包含从离子电导率测量得出的Li+活性项来描述。这些结果表明,像Li+的溶剂化能不仅受溶剂的调控,还受相应阴离子的调控,在相应的阴离子中,较高DN的阴离子也增强了Li+的溶剂化。
Li+的溶剂化吉布斯自由能受其配位环境的影响,光谱技术表明,配位环境由4个配位位点组成,来自溶剂,或者阴离子和溶剂的组合。拉曼光谱和傅里叶变换红外光谱(FTIR)是探测离子溶剂化环境的有力手段,因为溶剂-离子或离子-离子相互作用会改变分子振动的频率。例如,在纯1,2-二甲氧基乙烷(DME)的800–900 cm−1范围内的拉曼光谱显示,在≈821和≈849 cm−1处的两个宽峰,对应低聚醚中CH2的摇摆振动和C–O–C拉伸振动的混合模式(图2d)。在添加锂盐后,在≈870和≈876 cm−1处出现了新的谱带,这表明Li+-DME发生相互作用,其中这些新峰可以归属于环氧乙烷链(C−C−O)与反式(t)、gauche-plus(g)和gauche-minus(g′)序列构象组合的振动。但是,这些自由和Li+配位溶剂的拉曼散射系数几乎不变的,可以通过峰的去卷积得到Li+配位溶剂分子数的定量信息。
从这些研究中,作者经常观察到每个Li+有≈4个配位配体,对于每个溶剂有一个配位的溶剂(如二甲基亚砜DMSO和碳酸亚乙酯EC),对应于每个Li+周围的4个溶剂分子。通过拉曼分析,无论阴离子的DN多高,高DN溶剂DMSO都保留了≈4.3个配位每个Li+的溶剂氧原子。另一方面,较弱DME的溶剂配位可以越来越多地被阴离子取代,如图2e所示,DN从TFSI−的4.3个O/Li+(DN=5.4 kcal/mol)增加到TFA−的1个O/Li+(DN≈34 kcal/mol)。因此,参与Li+的第一溶剂化壳层是溶剂的给电子强度和阴离子的给电子力之间的竞争,其中较高的DN物种将优先与Li+相互作用。
此外,电喷雾电离质谱(ESI-MS)和核磁共振(NMR)等技术可以提供更加详细的溶剂化鞘信息。不幸的是,7Li NMR化学位移(以及13C和1H等其他原子核的化学位移)并不仅仅由有效核电荷调控,其中原子核周围的电子密度越小,就会导致向低场位移。然而,化学位移也会受价电子的总轨道角动量影响,其中由于电子供给减少而减少的轨道角动量会引起上场位移。因此,7Li、13C和1H NMR化学位移的观测结果应该使用其他技术的证据来支持。
2.溶解度对电池反应的影响
在锂离子电池中,Li+离子是唯一可溶于电解液的活性物质,而石墨负极和锂金属氧化物正极在其充电和放电状态下基本上都不溶。对于图3a中的不溶性反应物不溶性产物,作者将这种反应表示为I–I(Insoluble-Insoluble)。虽然这种结构简单的(脱)嵌入(I–I)反应产生了锂离子电池的高倍率和长循环寿命,但由于需要过渡金属离子来稳定脱锂正极,电极材料在锂化和脱锂状态下都保持稳定限制了电池的比能量。另一方面,Li–O2和Li–S等转化反应可以提供更高的比能,但需要电极材料在充电和放电过程中发生相变。例如,在Li–O2电池放电期间,气态O2可以被还原以形成不溶性固体过氧化锂导致S–I(Soluble-Insoluble)反应。此外,超氧化物锂(LiO2)等中间体也可溶解在电解液中,导致S–(S)–I反应(soluble-Insoluble–Insoluble)。在Li–O2和Li–S电池体系中,即使是很小的溶解度≈10−4 m也可能是显著的。
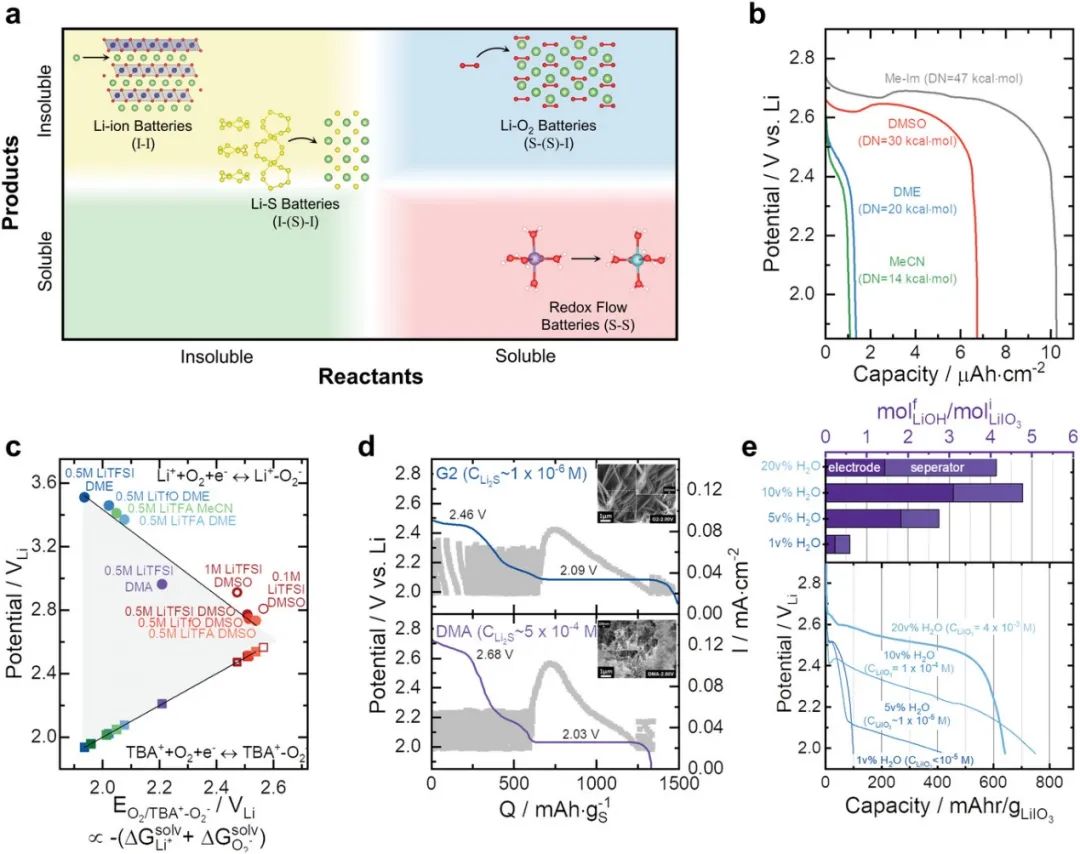
图3. 根据反应物、中间体和产物是可溶的还是不可溶的,对不同的电池化学成分进行分类。例如,S–(S)–I指可溶性反应物S–(可溶性中间体I)–不溶性产物S。
3.通过溶剂化相互作用改变物种的反应性
电解液种类的反应性决定了电池性能的关键方面,如循环寿命。例如,正极处的电解液分解会显著导致Li–O2电池的循环寿命变差。Feng等人的工作为Li–O2电池环境中溶剂的一些关键分解机制提供了一个框架,包括氢提取、去质子化、亲核攻击和电化学氧化,并确定了溶剂结构中哪些官能团可能容易受到这种分解机制的影响。
利用这个框架,开发了基于磺酰胺的电解液,它们对所有这些分解途径以及单线态氧的攻击都是稳定的。除了增强溶剂(电)化学稳定性的合成策略外,电解液内的分子间相互作用也会显著影响物种的反应性,从而提高溶剂的稳定性,但也能调节水等反应性添加剂。
3.1通过阳离子配位增强溶剂(电化学)稳定性
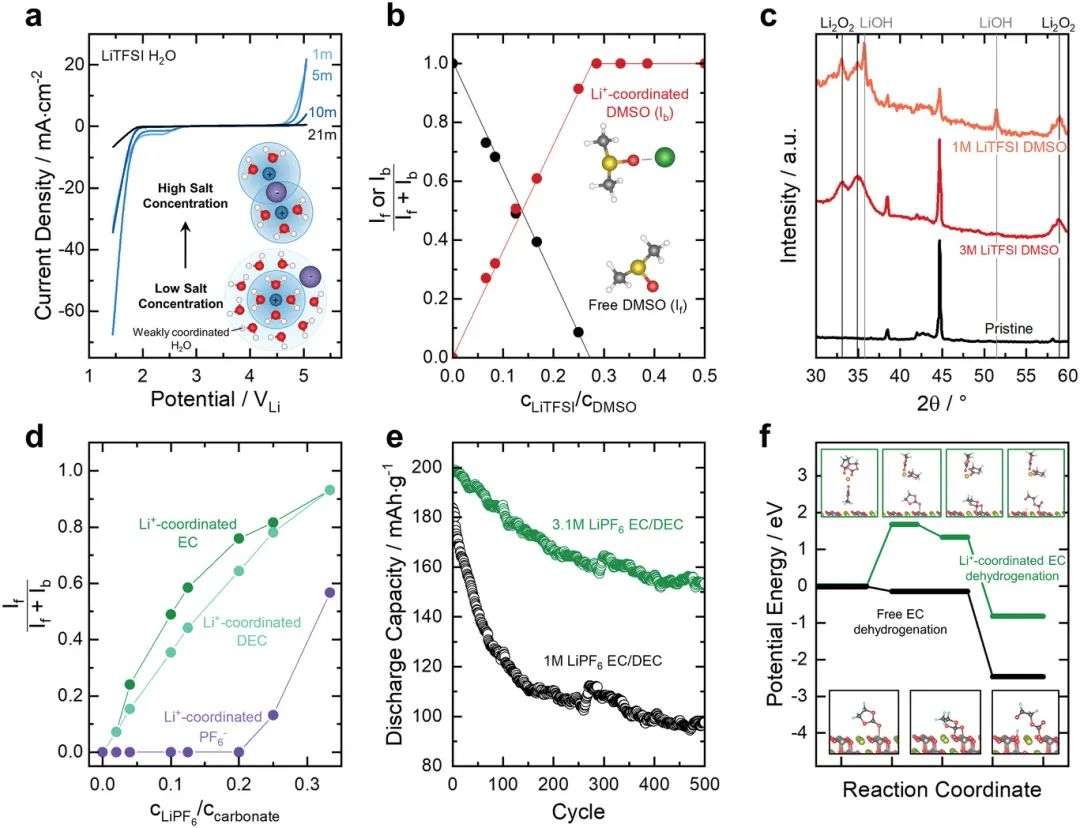
图4. a) 描绘不同盐浓度的LiTFSI 水系电解液的电化学稳定性窗口的循环伏安图。显示了盐浓度增加对水和Li+离子配位环境的影响的示意图。b) 拉曼光谱中游离和Li+配位的DMSO分子的比例,作为LiTFSI盐与二甲基亚硫的摩尔比的函数。c) 原始电极的X射线衍射(XRD)以及在1m LiTFSI和3m LiTFSI中放电后的电极。在更高浓度的3m LiTFSI-DMSO(其不具有游离DMSO)中,不存在DMSO分解以形成LiOH。d) 拉曼光谱中Li+-配位EC、DEC和PF6−的比例,作为LiPF6盐与EC/DEC溶剂摩尔比的函数。e) NCM811电池在1m LiPF6 EC/DEC和3.1m LiPF6 EC/DEC中的循环稳定性。f) 自由(黑色)和Li+配位(绿色)的EC脱氢的DFT计算的势能图。
溶剂分子和电解液中阳离子之间的配位可以增强溶剂的(电)化学稳定性。改变阳离子配位溶剂分子比例的最简单方法是改变溶剂和盐的化学计量(即盐浓度)。例如,索鎏敏的工作显示,随着LiTFSI盐浓度的增加,水性电解液的电化学稳定性窗口显著增强(图4a)。H2O的理论电化学稳定性窗口仅为1.23 V,但当盐浓度显著增加到21 m(mol盐/kg溶剂)时,可以扩展到≈3.0 V。这些“高浓度电解液”(HCE),几乎所有溶剂分子都与阳离子配位,进一步显示了对含LiTFSI电解液中Al腐蚀的抑制,石墨与PC电解液的兼容性,甚至乙腈对Li的化学稳定性。二甲基亚砜与Li+离子配位时,其化学稳定性增强。
从拉曼光谱来看,Li+-配位的二甲基亚砜分子的比例随着盐与溶剂的摩尔比线性增加,直到该比例达到≈0.25(1 LiTFSI:4 DMSO),之后发现所有二甲基亚硫分子都是配位的Li+离子(图4b)。然而,当LiTFSI在DMSO中的盐浓度增加,使得所有DMSO与Li+配位时,DMSO的这种化学分解被抑制,消除了LiOH的形成。例如,通过XRD比较1 m LiTFSI DMSO和3 m LiTFSI-DMSO的放电产物,作者发现较低浓度的1 m电解液同时显示Li2O2和LiOH,而高浓度的3 m电解液(图4b中不含游离DMSO)仅产生Li2O2(图4c)。
3.2通过溶剂和离子相互作用改变的电解液反应性
电解液中添加剂(如H2O)的溶剂化环境可以显著改变它们的反应性。例如,Kwabi等人比较了在Li–O2电池放电过程中,向0.1 m LiClO4 DME和AcN电解液中添加5000 ppm H2O的变化。即使Li2O2可以与纯H2O化学反应形成LiOOH和LiOH,但当5000 ppm H2O与DME混合时,它不再具有反应性,并且Li–O2电池的放电将仅产生Li2O2。这样的结果可以通过考虑1H NMR测量中H2O的化学位移来理解,这可以提供有关H2O酸度的信息,其中较高的化学位移对应于酸性更强的H2O。
值得注意的是,DME中5000 ppm H2O的1H NMR化学位移≈2.5 ppm,与本体水的4.8 ppm相比,表明DME中水的pKa高于本体水(即水的酸性较低)。DME中H2O的酸性降低可以抑制其被Li2O2去质子化,防止LiOOH和LiOH的形成(图5a)。另一方面,在MeCN中用5000 ppm H2O放电的Li–O2电池产生LiOH,而不是Li2O2,表明溶剂中水的pKa是溶剂依赖性的,其中更强的溶剂-水相互作用(即H2O和相邻溶剂分子之间更强的偶极-偶极相互作用)可能有助于水的去质子化。
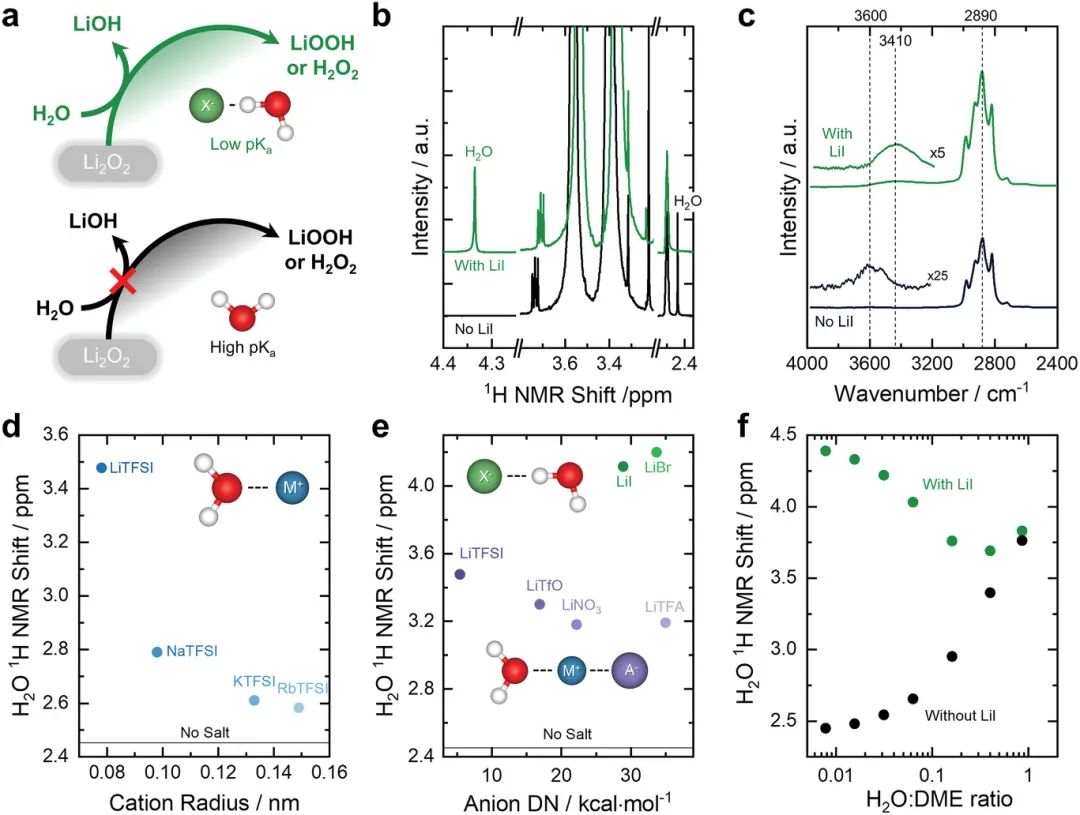
图5. a) 示意图显示,当向电解液中添加H2O由于其溶剂化环境而具有低pKa时,它可以容易地被Li2O2去质子化以形成LiOH和H2O2或LiOOH,而当它具有高pKa的时候,它不能被Li2O2。b) 具有1000ppm H2O和0.3m LiI以及1000ppm H2O的DME溶液的1HNMR光谱。纯H2O在T=298K下的1H NMR信号为4.8ppm。c) 纯DME与5000ppm H2O和0.3m LiI DME 5000ppm H2O的FT-IR光谱。d) 0.25 m H2O+0.2 m MTFSI DME溶液的H2O 1H NMR化学位移作为阳离子离子半径的函数,其中m = Li,Na,K,Rb等的离子半径。e)0.25 m H2O+0.2 m LiA DME溶液的H2O 1H NMR化学位移作为函数阴离子供体数,其中A=TFSI、OTf、NO3、TFA、I和Br。f) 对于含和不含LiI的H2O:DME混合物,作H2O:DEM比例H2O 1H NMR化学位移的函数。
离子配位可以促进非质子电解液中水添加剂的去质子化。作者与Tułodziecki等人之前的工作表明,基于1H NMR和FTIR测量,I−离子的存在可以显著改变DME中水的去质子化能。例如,作者发现DME中1000 ppm H2O的化学位移可以从没有盐的2.44 ppm显著位移到0.3 m LiI的4.34(图5b)。伴随着1H NMR中H2O化学位移的变化,电解液中H2O的O–H伸缩带从没有盐的3600 cm−1移动到存在LiI时的3410 cm−1,这与水的OH键减弱和更容易的去质子化进一步一致(图5c)。H2O在DME中与LiI的去质子化能降低,使Li–O2电池的放电产物从不含LiI的Li2O2变为含LiI的LiOH,这是由Li2O2对水的去质子化促进的。
在之前未发表的后续工作中(有关实验细节,请参阅支持信息),作者进一步表明阳离子和阴离子都可以改变DME中H2O的去质子化。例如,将TFSI−盐与尺寸越来越大的碱金属阳离子一起添加,导致H2O 1H NMR化学位移(酸度降低)从Li+的3.48 ppm降低到Rb+的2.58 ppm(图5d),这表明具有更高Lewis酸度的较小阳离子可以更强烈地配位水。此外,卤化锂(如LiBr和LiI)显示出比其他Li盐高得多的H2O 1H NMR化学位移和更高的H2O酸度,这表明Br−和I−离子可能与DME中的H2O直接相互作用,而其他阴离子仅用于调节Li+活性(图5e)。
3.3 Li+溶剂化对石墨和锂金属电极的影响
Li+的溶剂化环境可以使Li+可逆地嵌入石墨中,并影响SEI的组成和由此产生的Li金属负极的可逆性。商用锂离子电池中的石墨负极能够可逆插层,部分原因是电解液中存在EC溶剂。EC不仅有助于形成稳定的SEI,而且其在Li+的溶剂化壳中的存在促进了Li+离子的去溶剂化,导致电荷转移电阻降低。事实上,明军等人已经表明,即使在形成稳定的SEI之后,Li+离子的容易脱溶对于防止溶剂共嵌入和石墨剥落也是至关重要的,其中较弱的溶剂Li+相互作用有助于防止溶剂共插入。
对于Li金属负极(不受溶剂共嵌入的影响),SEI的组成不仅取决于电解液中物种的组成,还取决于这些物种是否存在于Li+的溶剂化壳中。这是因为Li+配位的溶剂分子的还原分解在热力学上比游离溶剂更优选,因为Li+配合位降低了溶剂的LUMO。此外,Li+溶剂化壳层中分子之间的相互作用可以进一步调节其还原稳定性。例如,Jiang等人已经表明,间苯三酚和1,3,5-三甲酰基氯苯酚等添加剂可以分别使Li+溶剂化壳中TFSI−阴离子的LUMO向上和向下移动。值得注意的是,虽然与Li+离子的配位可以改变溶剂和添加剂的反应性,但存在于Li+的溶剂化壳中的物种并不一定意味着它将有助于SEI的形成,也不影响由此产生的可逆性。
通过上面给出的例子,很明显,电解液内的分子间相互作用可以引起电解液组分反应性的显著变化。鉴于正极和负极对电解液(电)化学稳定性的重大要求,将基于分子间相互作用的设计策略与选择耐分解分子的合成策略相结合,对于实现能够提供长循环寿命的下一代电解液至关重要。此外,这种分子间相互作用可以提高对参与反应的添加剂(如水)的反应性的调控,从而能够开发具有更高能量密度的新型锂离子电池。
4.通过分子间相互作用增强离子电导率
传统液体电解液的离子导电性和低迁移数限制了电池的功率能力,从而限制了电池充电时间。在放电(或充电)过程中,低的Li+迁移数(会在整个电池和电极内引起浓度梯度,其中耗尽Li+离子的区域中的活性材料不再对器件容量有贡献,从而限制电池的倍率性能。因此,提高电解液的离子电导率和Li+迁移数以增加器件功率和缩短充电时间是非常有意义的。例如,利用室温离子电导率为25 mS/cm、tLi+≈1的固体陶瓷电解液,Kato等人开发了可以在18摄氏度下充电的所有固态电池,相当于在3分钟内充满理论容量的80%。在本节中,作者研究了分子间相互作用如何产生电解液的离子电导率和迁移数,为提高电池的倍率性能提供指导。
分子间相互作用可用于增加盐离子在低介电常数溶剂中的离解。溶剂对电解液离子电导率的影响已经得到了广泛的研究,其中离子电导率可以通过增加游离离子的浓度(通过增加盐浓度或增加盐的离解)或通过降低电解液的粘度来增加,这会导致更快的离子迁移率。然而,这些参数相互之间是相关的,一般具有更高介电常数的溶剂往往具有更高的粘度,而增加盐浓度也会增加电解液粘度。最近有一种在低介电常数电解液中增加盐离解的新方法,将大环阴离子结合受体(Cyanostar)添加到LiPF6- THF(ε0=6.7)电解液中。Cyanostar(CS)表现出尺寸选择性,并以2:1的三明治结构与PF6−阴离子强结合(图6)。通过在电解液中添加CS,5×10−3 m LiPF6, THF的离子电导率增加了一个数量级以上,从仅11 µS/cm增加到150 µS/cm(图6a)。
由于CS是一种尺寸选择性阴离子受体,作者进一步探讨了它对不同尺寸阴离子(如LiBr和LiTFSI)离子电导率的影响。5×10−3 m LiBr-THF(其中CS显示出与Br−和PF6−相似的亲和力)显示出与5×10–3 m LiPF6-THF相当的趋势,由于所有Br−被2 CS完全配位,在cCS/cLi=2处具有尖锐的弯头。另一方面,由于与CS的络合较弱,5×10−3 m LiTFSI-THF的离子电导率逐渐增加。通过对Li+–PF6−和CS–PF6–结合常数的UV–Vis和NMR测量对溶液形态进行测试,可以理解LiPF6-THF电解液离子电导率的显著增加。
这种配合物的形成表明,CS的加入增加了LiPF6接触离子对的解离,导致高比例的游离Li+离子和(CS)2PF6−复合物(图6b)。这些体积庞大的(CS)2PF6−复合物的形成(图6c)也减缓了它们相对于游离PF6−的迁移率,导致Li+离子的迁移数从纯LiPF6-THF电解液的≈0.5增加到0.8。虽然这种方法显著增强了低介电常数的THF中的盐离解,但CS的有限溶解度将最大离子浓度限制在<2×10−2 m。鉴于这种限制源于这种大环离子结合受体的溶解度,随后,研究者研究了操纵溶剂如何改变其与Li+离子、阴离子的相互作用及其自身相互作用,作为优化电解液离子电导率和迁移数的一种手段。
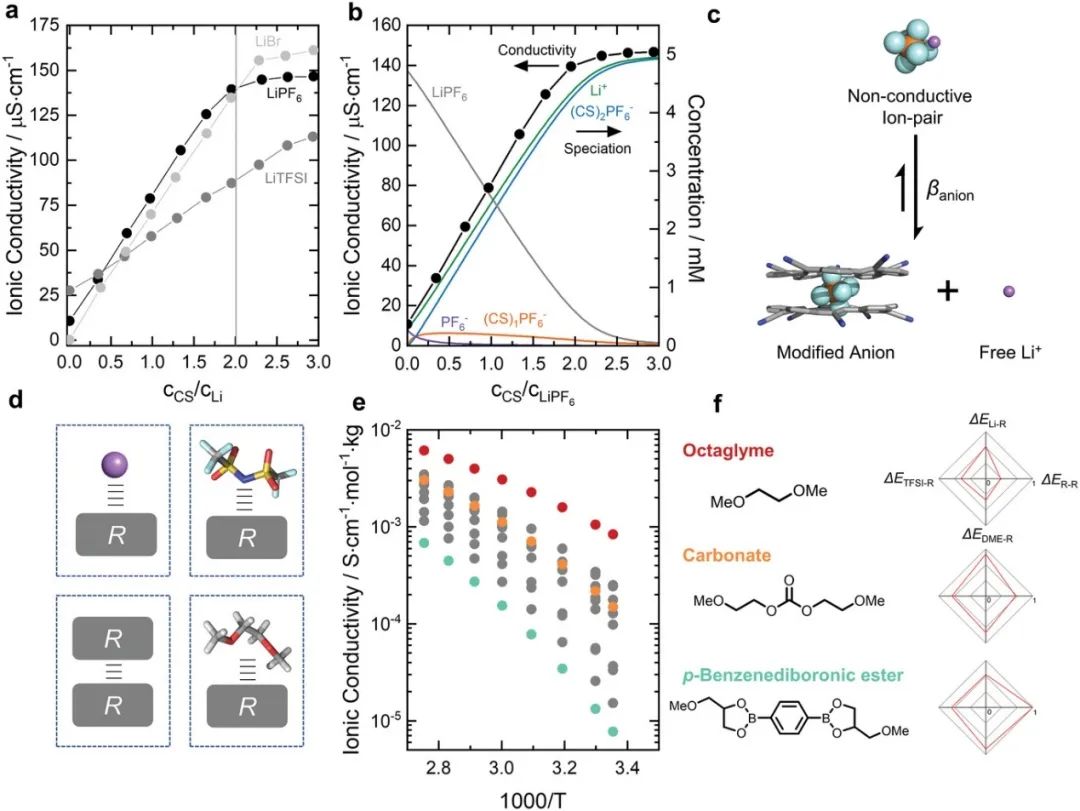
图6. a) 随着CS量的增加,THF中5×10−3 m LiBr、LiPF6和LiTFSI的电导率。b) 将CS添加到LiPF6中时形成的物质的电导率(黑点)和计算浓度(灰色、绿色、蓝色、橙色和紫色线)。该计算基于三平衡模型:LiPF6的离子配对(log Kip=4.9)、CS·PF6−1:1结合(log K1=5.3)和CS2·PF6–2:1结合(logβ阴离子=13.5)。c)离子对的超分子阴离子修饰方案。β阴离子是CS:阴离子复合物的总缔合常数。d) 使用DFT计算获得了气相中每个取代基与Li+、TFSI−、DME及其自身(自缔合)的成对相互作用能(ΔEi-R),并用于预测粘度和离子电导率。e) OEG-LiTFSI电解液的温度依赖性摩尔离子电导率(LiTFSI,Li/O=1/12)。f) 来自图e的三种化合物的化学结构和归一化相互作用能。
仅基于溶剂Li+、溶剂阴离子和溶剂-溶剂相互作用就可以相对较好地预测离子电导率。研究者们最近研究了一系列选择性修饰的低聚乙二醇(OEG)LiTFSI基电解液,使用了一系列具有不同Li–R、TFSI–R、DME–R和R–R相互作用的官能团(R)(图6d)。对12个不同的R基团使用DFT计算来计算每一个成对的相互作用能。值得注意的是,通过拟合一个非线性回归模型,该模型编码了Arrhenius温度依赖性和每个取代基的4 DFT计算的成对相互作用能,可以预测所得电解液的温度依赖性离子电导率(R2=0.98)和粘度(R2=0.99)。
这种非线性回归模型甚至可以预测训练集中未包括的3种化合物的离子电导率和粘度。这非常有意义,因为它们表明,即使是复杂的宏观性质,如离子电导率和粘度,最终也由电解液内的简单分子间相互作用调控。在作者的电解液系统中,R–R相互作用能(即官能团之间的偶极-偶极自相互作用)在确定所产生的离子电导率方面最为关键,因为强的R–R交互作用导致粘度显著增加和离子迁移率降低。例如,通过将取代基从八甘醇二甲醚、碳酸酯和对苯二硼酸酯改变来增加R–R相互作用能,导致室温下的摩尔离子电导率分别从8×10−4 S cm−1mol−1 kg−1降低到1×10−4 S cm−1mol−1 kg−1,再降低到8×10–6 S cm−1mol−1 kg−1(图6e,f)。
总结与展望
这篇mini综述中,作者展示了电解液中分子间相互作用对电池性能的巨大影响,如放电速率、放电容量和循环寿命。作者考虑了反应物和产物溶解度对电池反应的作用,表明即使溶解度<0.1×10−3 m,也可以实现溶解-沉淀反应途径,并且溶解度强烈影响放电过电位和容量。电解液组分之间的分子间相互作用显著影响了它们的反应性,导致高浓度电解液中溶剂(电)化学稳定性显著增强,水的去质子化能可广泛调节。最后,电解液的离子电导率和迁移数最终来源于电解液内的分子间相互作用,其中阴离子结合受体可以增强盐的离解,而减少溶剂分子之间的自相互作用可以降低粘度并增加离子迁移率,从而提高电池的倍率性能。
审核编辑:刘清
-
电解液
+关注
关注
10文章
846浏览量
23092 -
DFT
+关注
关注
2文章
224浏览量
22704 -
固态电池
+关注
关注
10文章
695浏览量
27771 -
锂金属电池
+关注
关注
0文章
135浏览量
4311
原文标题:MIT邵阳院士AEM mini综述:通过溶剂化作用调控电解液性质和氧化还原反应
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
贴片电解电容正负极判断方法
镍氢电池的电解液是什么
新宙邦拟在美国投建10万吨/年电解液项目
新宙邦美国路易斯安那州碳酸酯溶剂和锂离子电池电解液项目启动

非质子型弱配位电解液实现无腐蚀超薄锌金属电池

弱溶剂化少层碳界面实现硬碳负极的高首效和稳定循环

锂离子电池生产过程中湿度控制的重要性

锂电池电解液如何影响电池质量?锂电池电解液成分优势是什么?
无机锌盐中非质子性极性溶剂适用原则的深入分析!






 工商网监
工商网监
评论