 本源量子与复旦大学合作在分子晶体结构预测上获新进展
本源量子与复旦大学合作在分子晶体结构预测上获新进展
近日,本源量子研发团队和复旦大学的张俊良教授团队合作,利用量子叠加态的并行计算能力设计出新的分子晶体结构预测算法,证明了量子计算可以帮助化学家们用比传统建模方法更精准的方式,来预测晶体的分子结构。药物的理化性质与药物分子的结构和分子晶型息息相关。在分子晶体中,除了晶胞的结构,分子的取向与产生的偶极协同作用也会影响晶型,而晶型的数量会随着分子取向以及分子数目呈指数关系增长,因此从分子的微观性质预测最终可能的分子晶体结构是极其困难的【1】。为了更好地解决这一难题,研发团队展开了相关研究。01使用量子算法进行分子晶体结构预测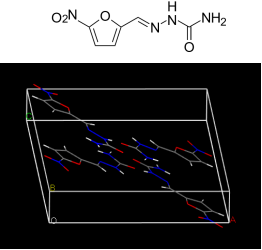 分子晶体结构预测的主要难点就在于需要对大量不同晶体结构并行搜索,传统计算无法满足大规模计算要求,而量子计算利用量子叠加态的原理,在模拟该问题的求解具有天然的优势,且随着问题规模的扩大,量子计算机的计算效率可以远超过传统计算机。
分子晶体结构预测的主要难点就在于需要对大量不同晶体结构并行搜索,传统计算无法满足大规模计算要求,而量子计算利用量子叠加态的原理,在模拟该问题的求解具有天然的优势,且随着问题规模的扩大,量子计算机的计算效率可以远超过传统计算机。
在分子晶体中,范德华力维系了晶体结构,而静电力(偶极作用)则决定了分子的方向。一个晶胞内有m个分子,每种分子就有n个方向,则由分子取向决定的分子结构就有m^n种。传统的周期性从头计算程序(VASP)可以提供足够精确的晶体结构和晶格能,但由于其高昂的计算耗费无法满足大规模搜索的要求。且简单分子力场的方法缺少对偶极协同作用的描述,0.5kcal的二体误差在复杂晶体计算中可能就被放大为100 kcal。此外分子力场通常不具备普适性,针对特定体系需要重新拟合和优化,因此整个过程非常复杂。本源量子团队利用量子近似优化算法(QAOA)对全局最小偶极能量和的选取进行了平方级加速,通过将问题编码为组合优化问题,为解决分子晶体结构预测难题铺平了道路。02XY混合层加速量子算法学习效率
在解决分子晶体结构问题时,为满足一个分子只有一个取向的限制条件,传统QAOA算法会使用增加惩罚项的方式,给不满足限制条件的解所代表的能量添加一个较大的数,从而筛选出符合限制条件的解,因此在QAOA搜索的过程中,会浪费大量资源,且如何选择合适的惩罚项也是一大难题。
为解决这类问题,本源量子团队采用了一种被称作量子交替操作算法【2】的QAOA变种,通过选择合适的初始量子态和量子操作,保证了最后结果一定落在可行解空间中,而无需添加惩罚项。这一方法不仅摆脱了超参数选择的困难,也极大提升了优化效率。
研发过程中,我们使用并改进了论文【3】中的方法,在不使用额外量子比特的基础上成功构造出了需要的最初量子态。通过XY混合层,我们仅仅使用了两层QAOA线路,就以100%的最优解概率在nitrofurazone晶体数据中成功得到了与经典遍历方法相同的结果。
研究人员还额外对比了XY混合层的两种实现方式(全连接complete、环形ring)与传统QAOA混合层(X)的表现,上图中横轴为量子门个数。可以看到,在相同的量子门数量时,XY混合层的效果要远好于传统的QAOA算法。
未来,本源量子团队与复旦大学将在该领域继续展开合作探索,通过对参数优化方式、编码方式、量子线路的排布等技术研究,实现量子算法在含噪声量子芯片上的落地,将进一步提供在线SAAS药物设计初筛、药效评估等模块,实现药物分子晶体结构在线预测和晶格能预测等功能,推进量子计算在生物医药领域的应用落地。
【1】Crystal Structure Prediction (CSP) Blind Tests
https://www.ccdc.cam.ac.uk/Community/initiatives/cspblindtests/
【2】HADFIELD S, WANG Z, O’GORMAN B, et al. From the Quantum Approximate Optimization Algorithm to a Quantum Alternating Operator Ansatz[J/OL]. Algorithms, 2019, 12(2): 34. DOI:10.3390/a12020034.
【3】Wang, Zhihui, et al. "XY mixers: Analytical and numerical results for the quantum alternating operator ansatz." Physical Review A 101.1 (2020): 012320.
-
量子计算
+关注
关注
4文章
1105浏览量
34966
发布评论请先 登录
相关推荐
纳芯微与复旦大学微电子学院合作研发成果亮相JSSC
天合光能与复旦大学共建先进光伏技术校企联合实验室

深度了解SiC的晶体结构

芯片和封装级互连技术的最新进展
上海光机所在提升金刚石晶体的光学性能研究方面获新进展
本源量子与广东联通签署战略合作协议

布局集成光量子计算!本源量子和硅臻芯片达成战略合作
本源量子与中国联通签署战略合作协议,共绘量子通信新蓝图
硅臻与本源量子达成战略合作协议
中国科学技术大学科研团队取得量子计算研究新进展
晶能光电与复旦大学合作研究用于可见光通信的红色发射微型发光二极管

基于量子干涉技术的单分子晶体管问世
从原子到超级计算机:NVIDIA与合作伙伴扩展量子计算应用
嘉善复旦研究院与复旦大学研发全无机钙钛矿集成光科技
清华大学在电子鼻传感器仿生嗅闻方向取得新进展





 工商网监
工商网监
评论