 揭秘锂氧电池充电慢的根本原因
揭秘锂氧电池充电慢的根本原因

01
导读
虽然锂-空气电池能够提供高的能量密度,但在放电过程中形成的绝缘Li2O2阻碍了接下来的再充电过程。氧化还原介质(RM)能够促进Li2O2氧化。低充电电压下的快速动力学对于实际应用是十分必要的,但尚未实现。
02
成果简介
近日,Nature Chemistry上发表了一篇题为“Why charging Li–air batteries with current low-voltage mediators is slow and singlet oxygen does not explain degradation”的文章,该工作研究了RM氧化Li2O2的机理。结果表明限速步骤是Li2O2外层单电子氧化为LiO2,遵循Marcus理论。第二步以LiO2歧化为主,主要形成三线态O2。单线态O2的产率取决于RM的氧化还原电位,而与电解质降解无关。该机制解释了为什么目前的低压介质(<+3.3 V)不能提供高的充电速率(最大速率在+3.74 V),并提出了重要的介质设计策略,以在更接近Li2O2氧化热力学电势(+2.96 V)下提供足够高的速率进行快速充电。
03
关键创新
该文章研究了氧化还原介质氧化Li2O2的机理。
04
核心内容解读
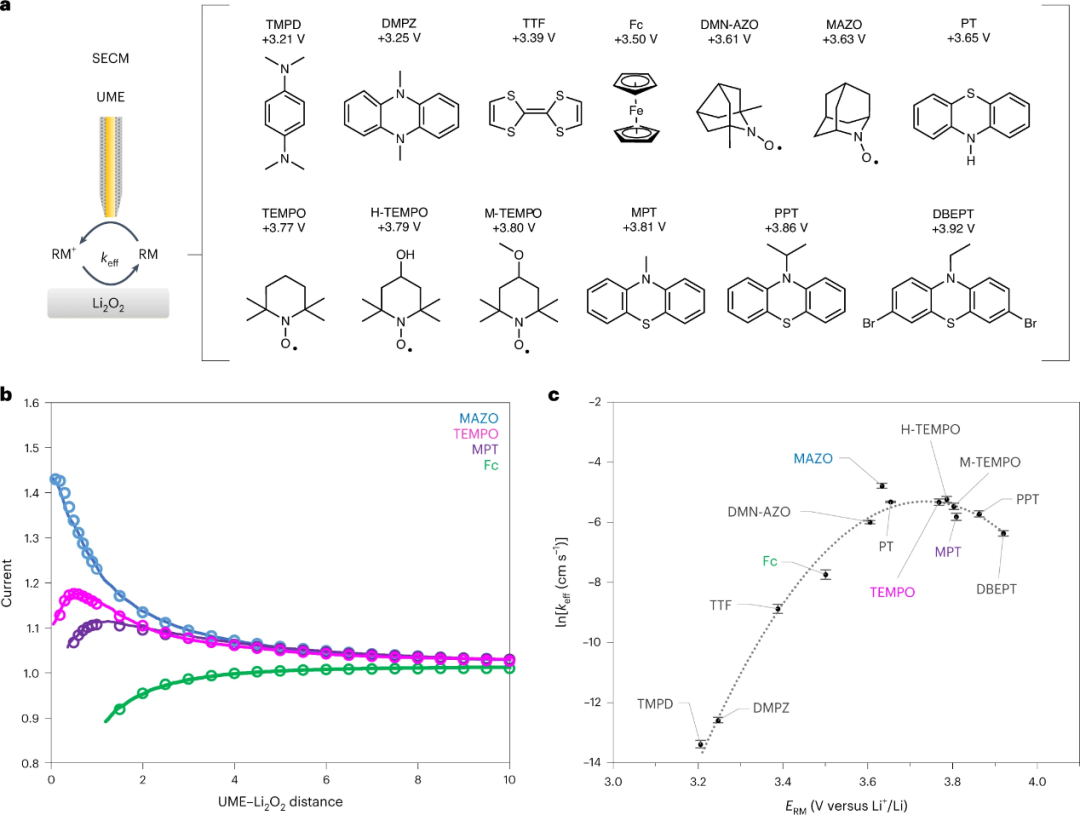
图1、a,在SECM设备中使用的UME示意图(左),以及所用RM的分子结构和ERM值(右)。b,绘制的RM的Li2O2的SECM逼近曲线。c, 在0.1 M LiTFSIin四乙二醇二甲醚中,lnkeff随ERM变化的Marcus曲线。@Nature
Marcus理论解释RM动力学
为了研究Li-O2电池在充电过程中通过RM(式(1))氧化Li2O2的机理,使用具有不同氧化还原电位(ERM)的RM(从+3.2 V到+3.9 V)对该反应的速率常数进行了测量(图1a)。
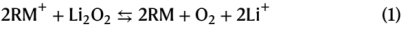
因此,得到了整个反应的有效速率常数(keff),该反应将由速率决定步骤主导。扫描电化学显微镜(SECM)广泛用于定量测量非均相电子转移速率常数。在本工作中,SECM采用图1a所示的装置来测量氧化的氧化还原介质RM+与Li2O2反应的keff。这可以通过绘制逼近曲线来实现,其中超微电极(UME)逐渐逼近Li2O2基底。RM在UME被氧化成RM+,然后与Li2O2基底反应,产生一个反馈回路,电流随距离而变化。图1b为所选RMs的反馈逼近曲线,其中在较低的UME-Li2O2距离下,较高的电流代表较高的keff。在Li2O2氧化的一系列步骤中(式(2)-(6)),keff与决速步骤的速率常数成正比。TMPD和DMPZ的keff值太低,无法通过SECM测量,因此使用压力传感的方法测量,其中Li2O2和RM+混合在气密室中,测量O2演变导致的压力变化。
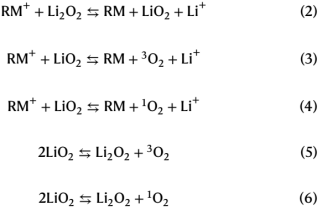
对于RM,将lnkeff数据绘制为ERM的函数,得到如图1c所示的火山状图。图1c中的曲线形状表明Marcus理论中电子转移速率常数(ket)的表达式如式(7)所示。
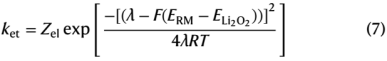
式中keff与ket成正比,ELi2O2为Li2O2的氧化还原电位,Zel为指前常数,λ为重组能,F为法拉第常数,R为气体常数,T为温度。λ在不同RM上的变化很小,因为它们的分子体积相似。将方程(7)拟合到图1c的数据中,可以看出keff随ERM的变化与Marcus理论预测的趋势一致。这意味着速率决定步骤是一个单一的外电子转移过程。
拟合值为:ELi2O2=+3.41 V,λ=0.322 eV, Zel=0.00495。Li2O2是由Li+配位的离散O22−分子组成的宽带隙固体。可以通过改变电解质环境来改变重组能(λ),其中含有四丁基铵(TBA+)盐的乙二醇二甲醚溶剂比碱金属盐的λ更大,这是由于它们的介电常数差异导致。
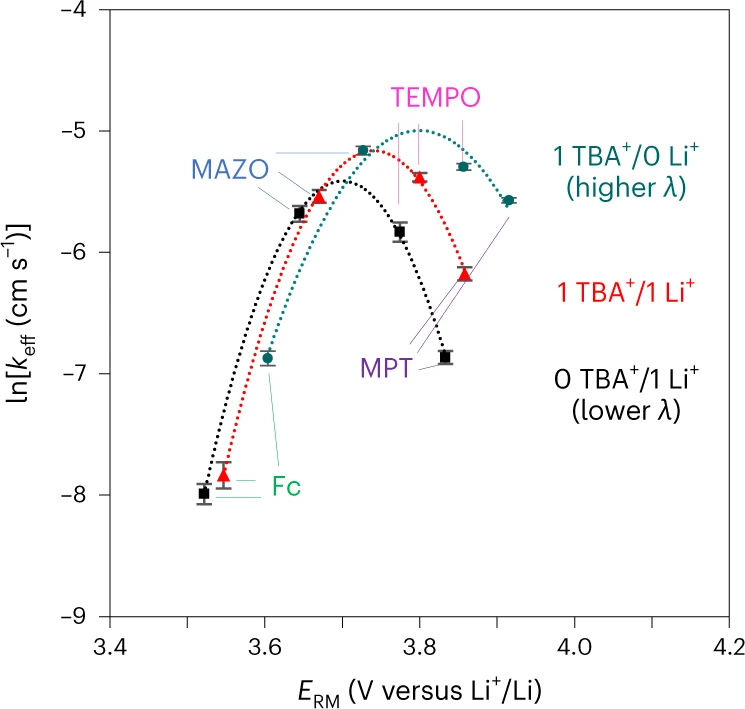
图2、当TBA+/Li+的总浓度保持不变时,lnkeff(RM+与Li2O2的反应)随ERM的变化曲线。@Nature
图2显示了lnkeff随各种电解质成分ERM的变化,其中Li+越来越多地被TBA+取代,导致Marcus火山图随着TBA+浓度的增加而向右移动。根据Marcus理论(方程(7)),火山的顶点(即最高氧化速率)发生在FΔE=λ处。因此,随着λ的增加,火山向更正的ERM移动。火山区域的扩大也与λ的增加相一致。这些结果进一步表明,RMs氧化Li2O2的动力学可以用Li2O2向RM+的外层单电子转移来解释。
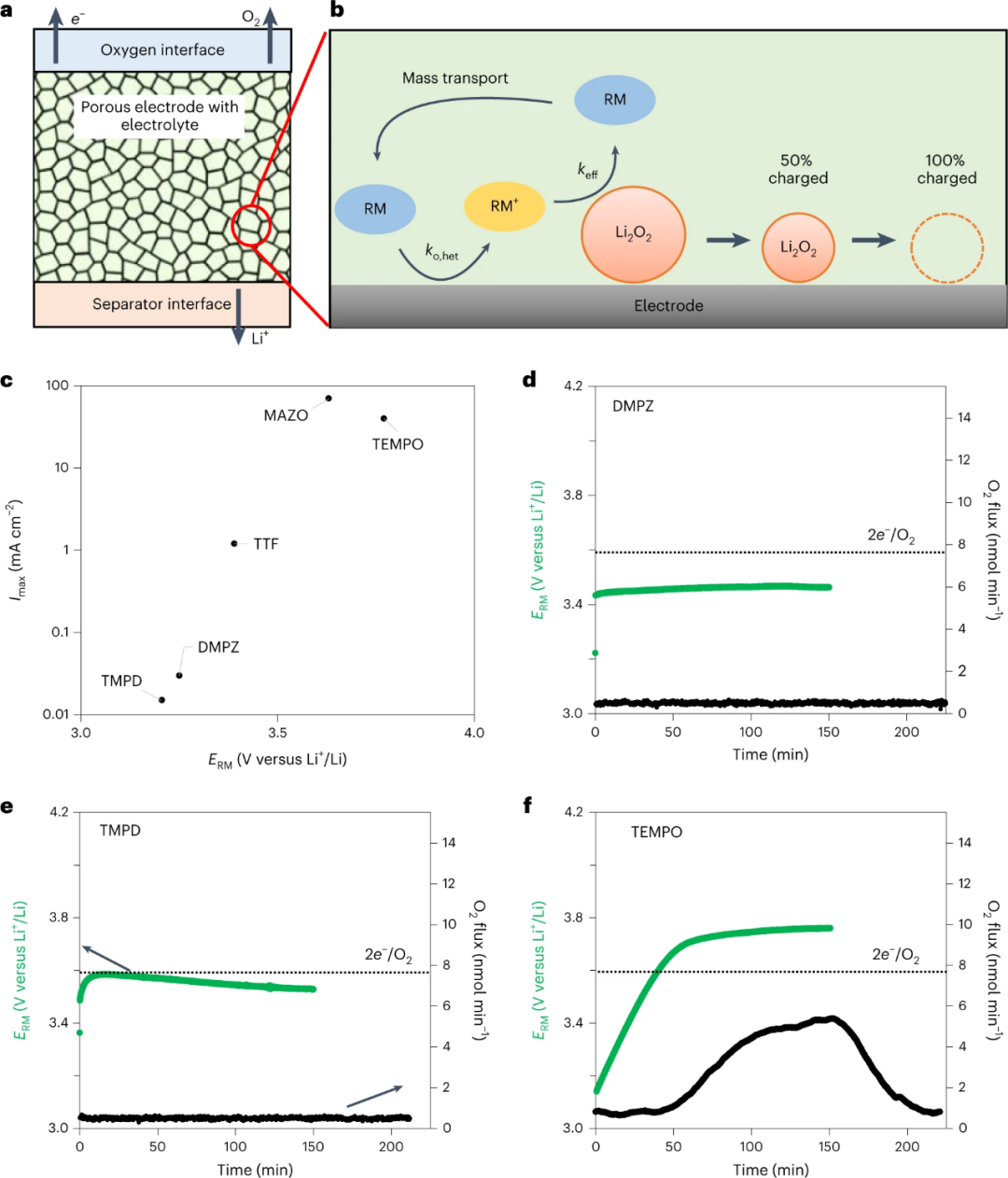
图3.a,用于计算具有RM的锂-空气电池恒流充电的多孔正极模型示意图。b、随着氧化的进行,颗粒尺寸减小。c,根据a中的正极模型,达到99.75%荷电状态的Imax随ERM的变化。d-f,预载Li2O2的Li-O2正极恒流充电曲线,使用1 M LiTFSI in 四乙二醇二甲醚电解质,添加10 mM DMPZ(d),10 mM TMPD(e)和10 mM TEMPO(f),并与在线质谱仪相结合,用于检测逸出的氧。@Nature
热力学上,对于电势>+2.96的任何介质,Li2O2都可能被氧化。然而,这并没有回答实际充电速率需要多少过电位的问题。接下来,使用如图3a、b所示的锂离子电池多孔正极模型,以及为每种介质确定的keff和ERM,模拟恒流充电曲线,以确定电池的充电速率极限。图3c显示了不同RM实现99.75%充电状态的最大充电速率(Imax)。RM在电极表面的氧化速率(ko,het)和RM在表面与Li2O2颗粒之间的传质速率很快,因此限制速率的步骤是Li2O2被RM+氧化的过程。图3c的结果表明,在极化到+4 V之前,较低电压的RM只能以较低的速率维持充电。例如,DMPZ和TMPD实现>99%充电容量的最大充电速率≤0.1 mA cm−2。相比之下,对于MAZO或TEMPO等高电压介质,可以在≥40 mA cm-2充电。
接下来,通过对连接到在线质谱仪的Li-O2电池充电,定性验证了该模型的结果。图3f显示,含有TEMPO的电池可以充电并产生大量氧气。相比之下,图3d,e显示,含有DMPZ或TMPD的电池只氧化RM,没有检测到任何氧气释放。对于后一种电池,虽然可以在电极表面直接氧化Li2O2,但电极是通过滴涂制备的,接触不良,加上介质动力学缓慢,电极中Li2O2含量相对较少,O2的析出被抑制在+4 V以下。
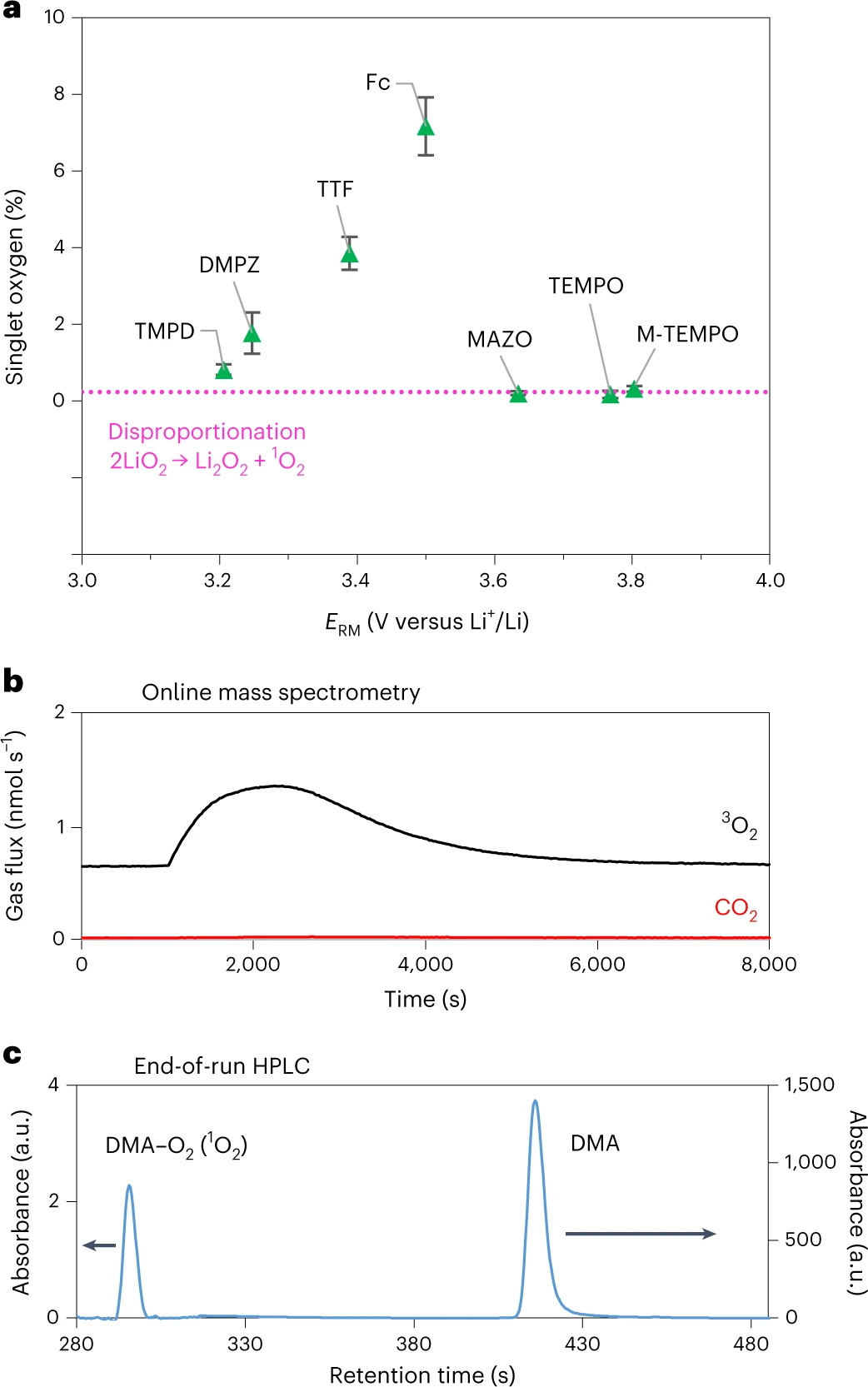
图4.a,单线态氧的产率(1O2/(1O2+3O2)百分比)随氧化还原电位(ERM)的变化,以及与LiO2歧化产率的比较(虚线)。b,从TEMPO+与Li2O2反应容器顶部空间取样的气体产物(黑色的3O2和红色的CO2)的在线质谱检测。c、通过对TEMPO+与Li2O2完全反应的液体溶液取样进行HPLC分析。@Nature
单线态氧的演化及反应机理
Li2O2首先氧化生成LiO2(式(2))。然后,LiO2通过两种途径,即LiO2氧化(式(3)和(4)或LiO2歧化(式(5)和(6)),以三线态(3O2)或单线态(1O2)的形式变成O2。总的Li2O2氧化反应应导致电荷/氧比例为e−/O2=2。然而,该值通常>2,这表明存在副反应。这种副反应归因于电解质溶液或碳正极受到反应性O2的攻击。之前的研究已经证明充电电压会影响1O2产率(即1O2/(1O2+3O2))。在这项研究之前,研究人员已经探索了用RMs给锂氧电池充电的O2产量。本工作通过将Li2O2与四烯胺中化学氧化的RM(RM+)混合来量化1O2产率,以了解RM+氧化在1O2产率中的作用。这个分析也使得能够确定式(2)-(6)中哪一个步骤限制了Li2O2的整体氧化速率。结果如图4所示。使用在线质谱法测量3O2的量,而1O2的量通过将其捕获在9,10-二甲基蒽(DMA)溶液中来测量,DMA是一种选择性化学陷阱,可与1O2快速反应形成DMA内过氧化物(DMA-O2),然后使用高效液相色谱法(HPLC)定量(图4c)。图4a显示了每种介质的1O2产率随ERM的变化。为了确定LiO2化学歧化直接产生1O2的产率,将超氧化钾(KO2)与含有Li+和18-冠-6-醚的四乙二醇二甲醚溶液混合,通过式(5)和式(6)诱导LiO2自发歧化。1O2产率为0.23%,如图4a中粉色虚线所示。图4a显示,对于ERM在3.2~3.5 V之间的RMs,1O2产率高于LiO2歧化产生的1O2产率,而对于ERM≥3.6 V的RMs,1O2产率是恒定的(与LiO2歧化的1O2产率一致),与电位无关。
由图1可知,Li2O2氧化的决速步骤是第一或第二单电子氧化,即Li2O2→LiO2或LiO2→1O2或3O2。在图4a中,LiO2歧化产生的多余1O2一定来源于第二电子转移,即LiO2氧化生成1O2(式(4))。对比图1中keff的变化趋势(即Li2O2氧化过程中限速步骤的有效速率常数)与图4a中形成的1O2比例变化趋势,可以明显看出,限速步骤不是RM++LiO2→RM+1O2+Li+,因为Li2O2氧化最大速率(图1)对应的电位与图4a中最高的1O2比例并不一致。事实上,在最大的keff下,仅通过歧化就可以形成1O2。根据类似的论点,可以排除RM++LiO2→RM+3O2+Li+是决速步骤。因此,限速步骤是Li2O2的第一电子氧化:RM++Li2O2-RM+LiO2+Li+(式(2))。图4a中O2的产率表明O2的主要产物是3O2,因此LiO2氧化或歧化的主要产物是3O2而不是1O2。图4a还显示,RM++LiO2→RM+1O2+Li+发生在3.2-3.5 V的ERM下,曲线的形状表明,RM介导的LiO2氧化也可能遵循Marcus关系。基于这一假设,可以预测3O2的能量比1O2低0.97 eV,即RM++LiO2→RM+3O2+Li+发生在<+2.96 V。这意味着在Li2O2被氧化的所有电位下,RM++LiO2→RM+3O2+Li+已经反应完毕。RM++LiO2→RM+1O2+Li+通过歧化途径争夺LiO2。随着RM电位的增大,Li2O2氧化生成LiO2的速率加快,LiO2浓度升高,RM+浓度降低,有利于LiO2歧化。简而言之,参与RM++LiO2→RM+1O2+Li+反应的LiO2和RM+的量不是一个常数。
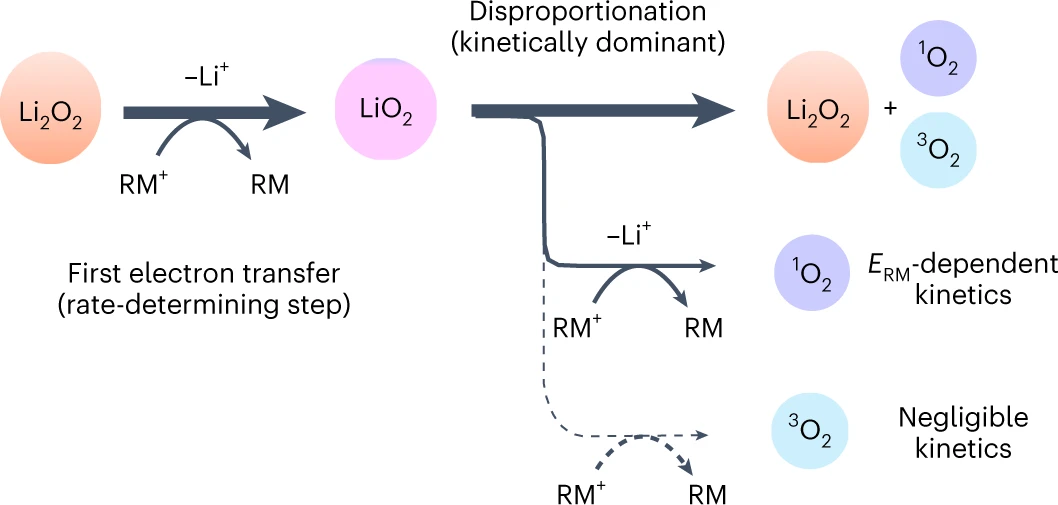
图5.RM促进Li2O2氧化反应机理示意图。@Nature
总之,Li2O2氧化的限速步骤是Li2O2的外层单电子氧化生成LiO2(式(2)),第二步是LiO2歧化生成3O2(式(5))。总体机理如图5所示。
05
成果启示
本文对Li2O2氧化机制的理解解释了为什么在低电势(<+3.3 V)下工作的介质,具有的良好往返能量效率,却不能提供足够高的充电速率。最优的RM是在低电势下充电的RM(也就是说,刚好足以在+3 V左右的热力学条件下驱动反应),并且具有快速的动力学和最少的反应性1O2形成。然而,在+3.7 V左右的ERM下才能获得最高的速率。考虑到限速步骤遵循Marcus动力学,对于接近ELi2O2的低电压RM,通过最小化重组能λ来降低活化能,有望实现快速的Li2O2氧化动力学(keff)。计算模型表明,氧化还原电位为+3.4 V的RM可以实现1 mA cm−2的充电速率。本文的分析表明,当ERM约为+3.45 V时,RMs的反应性1O2生成量达到最大值,但当ERM低于+3.2 V时,1O2的生成量接近基线值(即通过歧化生成的量)。因此,低电压RM也将使1O2生成量最小化。对于在产生1O2量最多的电位(约为+3.45 V)下工作的RMs,e-/O2比例接近2,这与在产生1O2量较少的电位下工作的RMs的e-/O2比例非常相似。此外,Li-O2电池充电过程中的降解和副反应在3.6 V以上最为显著,此时1O2释放最低。这些结果表明,在充电过程中,1O2可能不是副反应的主要来源。
审核编辑:刘清
-
锂电池
+关注
关注
263文章
8752浏览量
186497 -
电池充电
+关注
关注
9文章
507浏览量
76321 -
电解质
+关注
关注
6文章
839浏览量
21517
原文标题:牛津大学Nat. Chem.:揭秘锂氧电池充电慢的根本原因
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
锂金属电池革命:揭秘锂负极表面改性的最新突破与挑战

SGM41524A/SGM41524B:紧凑型开关式锂/聚合物电池充电器的卓越之选
深度解析SGM41524:紧凑型开关式锂/聚合物电池充电器
LTC1734:高效锂离电池充电器的设计与应用
LTC1732-4/LTC1732-4.2:高效锂离电池充电控制器的卓越之选
ADP5063:单节磷酸铁锂(LiFePO4)电池充电器的卓越之选
通过定制化充电协议提升初始无负极锂金属软包电池性能:机理与应用
晶振使用中常见问题与解决方法

如何用超级电容取代钛酸锂电池,破解蓝牙温度计欧盟出口认证与寿命难题?

技术深度解析:永铭锂离子电容如何实现无电池遥控器终身免维护

rt_sem_take卡住导致线程无法正常运行怎么解决?
技术深解|永铭超级电容如何解决新能源车碰撞断电车门锁死危机?

STM32G473 flash擦除时程序卡死的原因?
部分外资厂商IGBT模块失效报告作假对中国功率模块市场的深远影响




评论