 钾电与锂电电解质离子输运及热力学性质差异的根源!
钾电与锂电电解质离子输运及热力学性质差异的根源!
背景介绍
自2021年初以来,锂价格上涨了7倍多,这对锂离子电池(LIB)供应链提出了重大挑战。预测显示,到2030年可能出现严重的锂供应短缺。在代替锂离子电池的碱金属电池中,钾离子电池(KIB)比钠离子电池(NIB)有明显的优势,因为K+可以可逆地插入到石墨电极中。快速充电(~4C)对于电池来说也十分重要,特别是在电动汽车(EV)中,然而,传统的LIB倍率性能有限。KIB在倍率和功率方面可能比LIB有优势。早期数据显示,与LIBs相比,KIB的倍率性能有所提高,这表明KIB在非水系钾离子电解质中的传输速度更快。与Li+相比,K+尺寸更大,电荷密度更低,因此与溶剂分子的相互作用更弱,斯托克斯半径更小,这有望促进离子在电解质中的传输。虽然研究表明K离子电解质的倍率性能有所提高,但要全面了解K离子电解质的传质,只能通过全面准确地描述基本的离子传输和热力学性质:盐扩散系数(D)、阳离子转移数(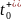 )、离子电导率(κ)和热力学因子(χM)来实现。然而,目前还没有研究完全表征了K离子电解质的离子传输和热力学性质。
)、离子电导率(κ)和热力学因子(χM)来实现。然而,目前还没有研究完全表征了K离子电解质的离子传输和热力学性质。
正文部分 01 成果简介
近日,牛津大学MauroPasta教授,研究了由双氟磺酰亚胺钾(KFSI)盐和1,2-二甲氧基乙烷(DME)溶剂组成的非水系K离子电解质溶液的离子传输和热力学性质,并将其与浓度范围0.25-2M的锂离子电解质(即LiFSI:DME)进行了比较。使用定制的K金属电极,证明了KFSI:DME电解质溶液比LiFSI:DME溶液具有更高的盐扩散系数和阳离子转移数。最后,通过Doyle-Fuller-Newman(DFN)模拟,研究了K∣石墨和Li∣石墨电池的K离子和Li离子存储性能。
02 图文导读
【图1】K电极制备方案示意图,以表征K离子电解质盐扩散系数(D),阳离子转移数(),离子电导率(κ)和热力学因子(χM)。与LiFSI:DME相比,KFSI:DME的D和较高,导致浓度梯度较低。
制备金属钾电极
本研究开发了一种K电极制备方法,以确保K金属具有足够的稳定性。第一步是在一个充满Ar的手套箱中熔化K金属,撇去漂浮在熔体上的杂质,然后淬火。在电池组装之前,将一个干净的K金属球体辊成金属片(厚度约0.6mm),冲孔,并使用微切割技术对表面进行抛光(图1)。
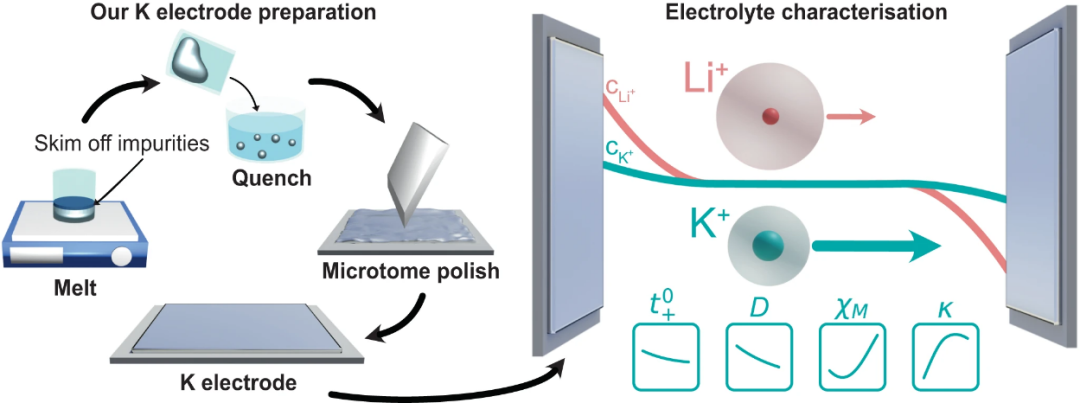
【图2】本工作制备的K电极与标准K电极组装的K∣∣K对称电池在20°C下,1M KFSI:DME中的稳定性。(a)静置8小时的平均初始OCV曲线。(b)在20°C下休息1小时后的阻抗图,插图为AFM高度图。Ar+溅射5,15和25min后K金属的XPS深度分布图(c)标准K电极的O1s, K 2p和C1s光谱和(d)本工作制备的K电极O1s, K 2p和C1s光谱。
图2a显示,与文献中报道的标准制备过程相比,本工作制备的K电极对称电池具有低的开路电压(OCV),表明表面稳定性和均匀性得到了改善。与标准方法相比,使用该制备方法制备的电池总阻抗大大降低(图2b)。图2b中插入的原子力显微镜(AFM)图进一步表明该方法能够实现表面均匀性,制备的K表面是平坦的,没有污染。X射线光电子能谱(XPS)和Ar+深度分析显示,用本工作中的方案制备的电极O1s光谱(图2d)由529.3eV处的KOH峰主导。由于K金属的高反应活性,这种KOH可能在XPS内形成,因为新鲜的K金属暴露,表明表面富含金属K。
相反,图2c中的氢氧化物峰强度相对较低,结合能为529.9eV,与NaOH一致。这表明在标准电极的表面只有有限的金属K可以利用。图2c中标准K电极的K2p XPS光谱显示,随着溅射深度的增加,K2p双峰面积减小,而图2d中本工作制备的K电极K2p双峰在溅射过程中强度保持不变。在图2c中,标准K电极表面还有一个C1s峰,表明有碳化物。因此,本工作的K电极制备工艺产生的富K电极表面具有更高的均匀性和更低的杂质元素。
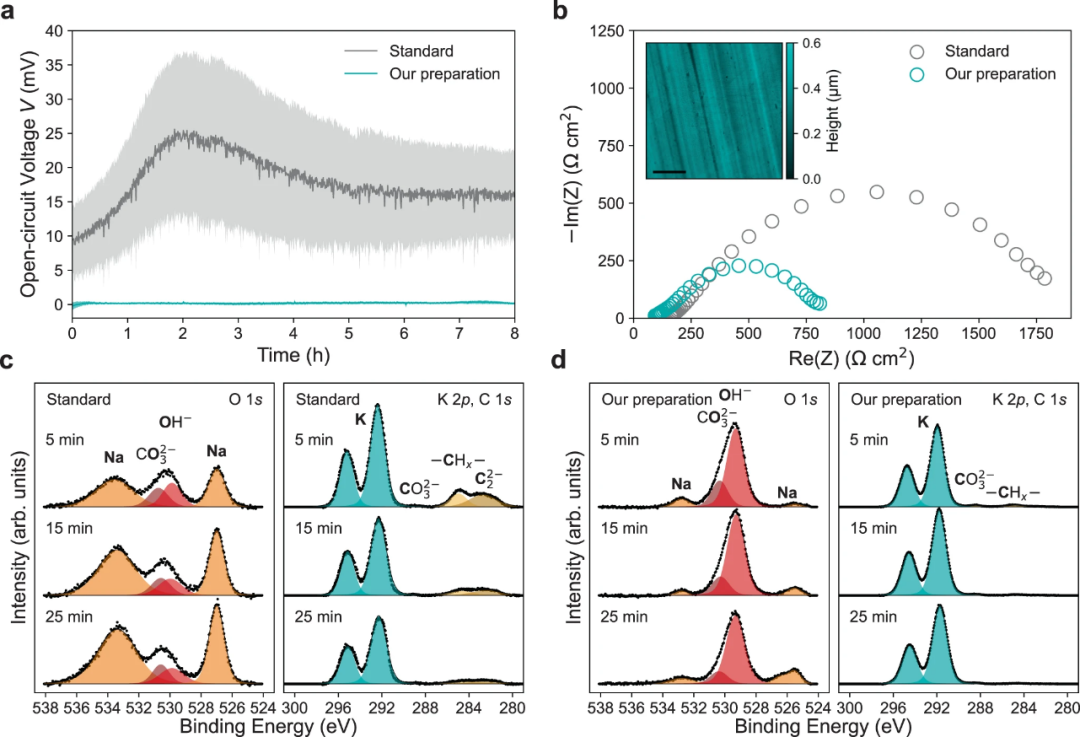
【图3】20℃时,KFSI和LiFSI在二甲醚中的转移数和离子电导率。(a)由希托夫实验测得的阳离子转移数。(b)用电导池测量的离子电导率κ。
迁移数
阳离子转移数为,是阳离子携带电流的分数。本工作采用希托夫法来表征迁移数。对一个大的对称电池施加极化,然后关闭两个旋塞形成三个隔离腔,提取溶液并测量其密度以确定浓度变化。通过公式(1)确定转移数。K离子和Li离子电解质盐的偏摩尔体积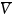 e用于确定。
e用于确定。

式中,Vchamber为阴极或阳极腔的体积,cf为实验后腔的浓度,c为中性腔的浓度,Ipulse为施加的电流,tpulse为脉冲持续时间。图3a显示了DME中KFSI和LiFSI的有效迁移数。结果表明,在较低的浓度0.25M时,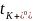 高于
高于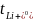 (分别为0.49和0.34),但随着浓度的增加,逐渐降低,在1.5~2M时仅略高于,两者呈现出逼近的趋势,说明较低的K+电荷密度抑制了浓度增加时离子-离子和离子-溶剂的相互作用。随着浓度的增加,从0.49左右下降到0.38,表明离子-离子和离子-溶剂相互作用的增加对K+的结合作用比FSI−更强。而对于Li+,在整个浓度范围内保持恒定。
(分别为0.49和0.34),但随着浓度的增加,逐渐降低,在1.5~2M时仅略高于,两者呈现出逼近的趋势,说明较低的K+电荷密度抑制了浓度增加时离子-离子和离子-溶剂的相互作用。随着浓度的增加,从0.49左右下降到0.38,表明离子-离子和离子-溶剂相互作用的增加对K+的结合作用比FSI−更强。而对于Li+,在整个浓度范围内保持恒定。
离子电导率
图3b显示,从低浓度到~1.5M, LiFSI具有较高的κ,此后κ显著降低。然而,KFSI的κ随浓度持续增加,并在2M处趋于平稳。这表明,与~1.5M以上的KFSI电解质相比,LiFSI电解质可能存在显著的物种间相互作用。1.5M以下的K离子电解质电导率低于Li离子电解质。由于KFSI的χM在较低浓度下比LiFSI更接近理想状态,表明离子-离子相互作用比Li+更少,因此在大多数浓度范围内,KFSI的离子电导率较低可能是由于KFSI盐解离度较低。
在高于1.7M的浓度下,LiFSI电导率的下降可能是由于Li+的离子-溶剂和离子-离子相互作用更大,因为其具有更高的电荷密度,Li+比K+能够拖拽更多的溶剂,因此具有更强的溶剂化作用,而Li+更强的库仑相互作用也导致更大的离子-离子结合,形成聚集体,增加了电解质粘度。在KFSI:DME中,只有浓度超过一定范围(>3M)才会形成聚集体。DME电解质的离子电导率较高,KFSI:DME在2M处的电导率为16mS cm-1,LiFSI:DME在1.5M处的电导率为15mS cm-1。
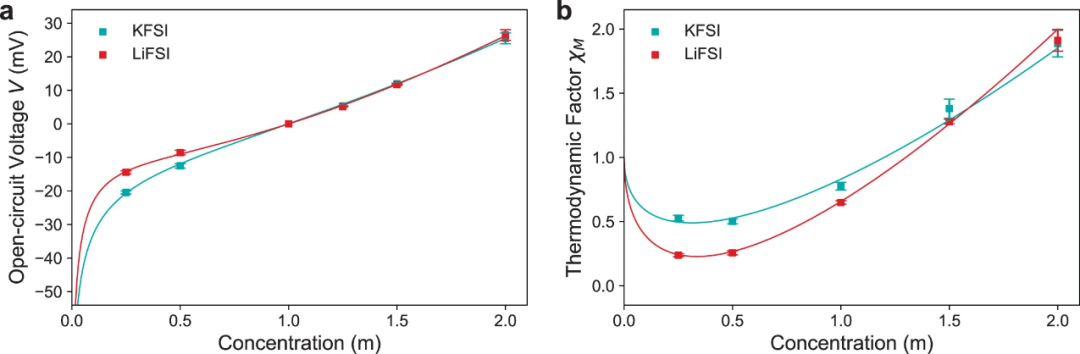
【图4】20°C下,KFSI和LiFSIin DME的浓度池和热力学因子数据。(a)浓度池开路电压V。(b)热力学因子χM。
热力学因子
热力学因子χM用于衡量电解质的非理想性,并解释了与能斯特行为的偏差,反映了盐的热力学活性如何随浓度变化。使用浓度池测量χM,其中在“测试”溶液和“参考”溶液之间测量开路电压。通过式(2),横跨浓度池的OCV变化V随摩尔浓度c的变化与热力学因子χM和迁移数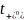 有关:
有关:
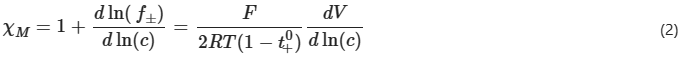
式中f±为平均摩尔活度系数,F为法拉第常数,R为气体常数,T为绝对温度。溶剂浓度c0和溶剂的偏摩尔体积0可以用来计算热力学因子。根据推导,将χM拟合到式(3)中给出的函数:

cm是摩尔浓度。A1和A2是拟合常数。OCV数据的拟合结果如图4a所示。图4b显示了热力学因子随浓度的变化情况。随着浓度的增加,第一库仑离子-离子相互作用降低了盐相对于DME的自由能,降低了盐活度系数,从而导致χM下降。随着浓度的进一步增加,离子-溶剂相互作用增加,导致DME越来越多地结合,降低了溶剂蒸气压,从而增加了盐活度系数和χM。结果表明,KFSI在较低浓度下的χM下降幅度明显小于LiFSI。这可以归因于以下两个因素。首先,与Li+相比,K+尺寸更大,因此电荷密度更低,导致K+的库仑离子-离子相互作用更弱,因此χM的减小较小。其次,从Debye-Hückel理论来看,在较低浓度下,DME中LiFSI和KFSI的χM下降梯度应该相等,因为它是由溶剂的介电常数ε决定的。由于Debye-Hückel理论假设电解质完全解离,较低的KFSI梯度进一步表明盐解离较差,因此在低浓度下其具有较低的离子电导率(图3b)。在较高浓度下,LiFSI的χM随浓度的增加也高于KFSI。这也是由于K+的电荷密度降低,导致与溶剂分子的相互作用减弱,因此与Li+相比,K+的溶剂化壳层更小。与Li+相比,较弱的K+溶剂化导致游离DME更多,因此KFSI的盐活度系数和χM随盐浓度的增加而降低。
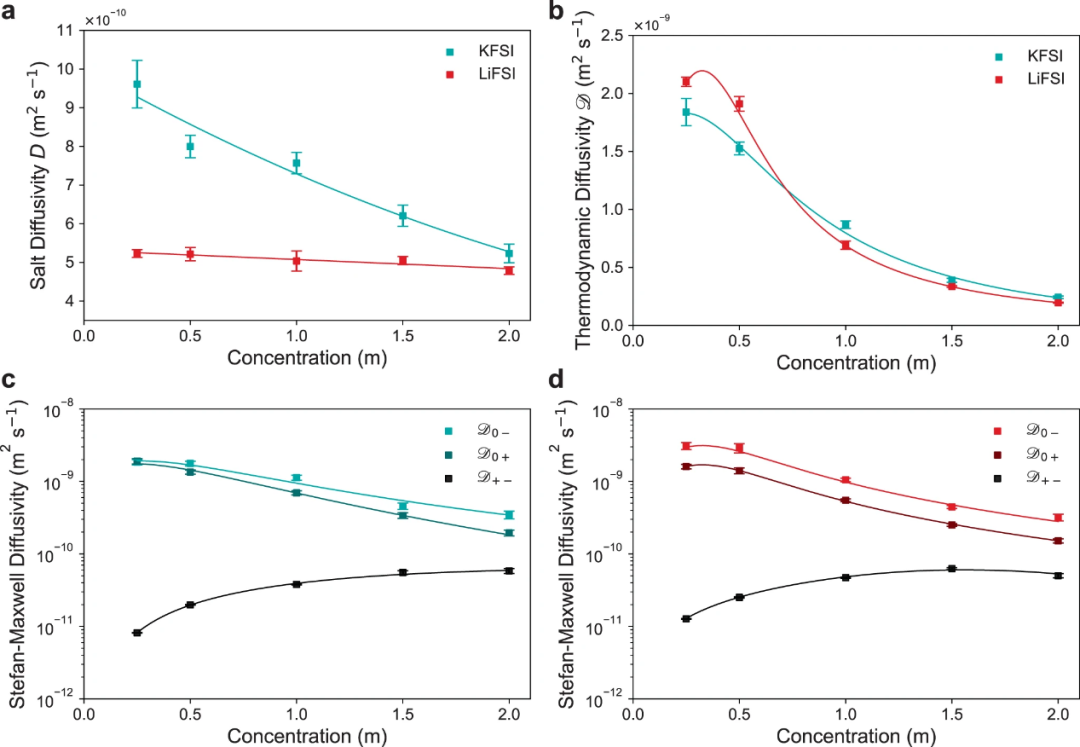
【图5】用稳态恒流限制扩散法测定KFSI和LiFSI在20℃二甲醚中的扩散系数。(a)盐扩散系数D。(b)采用实测的热力学因子计算热力学扩散系数D。(c)KFSI和(d)LiFSI的Stefan-Maxwell扩散系数Dij。
扩散系数
稳态极化和长期弛豫限制扩散被用来表征D,因为它比脉冲极化方法更准确,更不容易受到双层弛豫效应的影响。本工作设计了一个受限扩散池,并使用恒流极化来形成浓度梯度。扩散系数D可以通过式(4)确定。

式中,V为弛豫时测得的受限扩散池OCV,Ls为体相电解质厚度,t为时间,τdiff为特征衰减时间=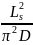 。
。
图5a显示,在20°C,所有浓度下KFSI的D都高于LiFSI。在1M处,DKFSI比DLiFSI高50%以上(分别为7.6× 10−10m2s−1和5.0× 10−10m2s−1)。这种差异在低浓度下最为显著,DKFSI几乎是DLiFSI的两倍。随着浓度的增加,DKFSI和DLiFSI之间的差异变得更小。DKFSI和DLiFSI都随着浓度的增加趋向于相似的值,表明较低的电荷密度似乎延迟了浓度增加时离子-离子和离子-溶剂相互作用。
图5b是用χM将D转化为热力学扩散系数D,反映了相对于盐化学势梯度的扩散系数而不是浓度梯度。D的初始增加是由于离子结合的增加导致,这是由于两个物种有效地合并为一个物种,从而降低了对单个物种运动的阻力。如χM所示,由于Li+的离子-离子相互作用大于K+,DLiFSI显示出更大的初始增加。在较高浓度下,DKFSI仅略高于DLiFSI,但表现出几乎相同的趋势,表明扩散行为差异与浓度梯度有关,而不是化学势梯度。
Stefan-Maxwell扩散系数根据驱动扩散的热力学力来描绘每种电解质物种相对于彼此的迁移率,从而更深入地理解扩散行为。KFSI和LiFSI电解质的Stefan-Maxwell系数分别如图5c、d所示。KFSI和LiFSI的系数相似,这是由于它们具有相似的热力学扩散系数。在整个浓度范围内,LiFSI和KFSI的溶剂-离子扩散系数D0+和D0−都减小了约一个数量级,表明DME对阳离子和阴离子的阻力随着盐浓度的增加而增强。在LiFSI的浓度范围内,D0−均高于D0+,表明与Li+与DME的相互作用相比,FSI−与DME的相互作用较弱。然而,对于KFSI,D0+和D0−更接近,在低浓度下几乎相同。这是由于在0.25M处K+的为~0.5。随着浓度的增加,D0−和D0+之间的差异增大。LiFSI和KFSI的离子-离子扩散率D+−比溶剂-离子扩散率大约低两个数量级,特别是在较低浓度下,表明两者的离子相互作用都有些显著。对于KFSI和LiFSI来说,D+−的最大值与它们的最大离子电导率相匹配,这表明在Li+浓度较低的情况下,由于缺乏游离DME,导致了更大的阳离子/阴离子相互作用。
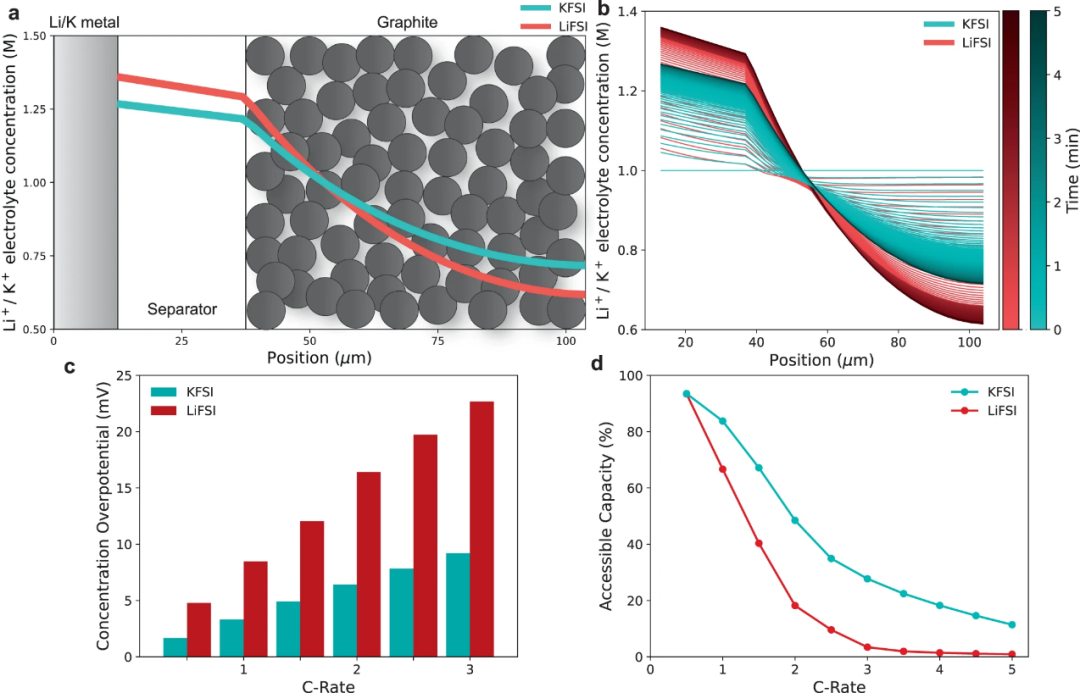
【图6】使用KFSI和LiFSIinDME电解质充电时,K∣石墨和Li∣石墨电池的DFN模拟。(a)模拟的金属||石墨电池在2C恒流充电5分钟后,Li+/K+电解质浓度梯度。(b)2C充电时Li+/K+电解质浓度梯度随时间的变化。(c)随着倍率的增加,金属||石墨电池的浓差过电位。(d)随着倍率的增加,金属||石墨电池的容量。
模拟
为了了解电解质传输和热力学性质差异对电池性能的影响,对采用KFSI和LiFSI:DME电解质的K离子和Li离子石墨半电池充电行为进行了Doyle-Fuller-Newman(DFN)模拟。图6a描绘了模拟的金属∣∣石墨电池。在2C(1C=3.28mAcm-2)充电5分钟后,KFSI电池与LiFSI电池相比,电解质浓度梯度降低。由于存在过电位,在此之后不久,LiFSI电池达到了0.01V的截止电压,低于此电压将开始发生锂金属电镀。图6b显示了在相同充电速率下随时间变化的浓度梯度,结果再次显示,KFSI电池的电解质浓度梯度减小。图6c显示,浓度梯度的减少导致浓差过电位降低(在充电速率1–3C下,KFSI比LiFSI电池低~60%),再次证明了和D的重要性。最后,图6d显示,KFSI电池可以在更高的充电速率下实现更高的容量。较高的锂离子浓差过电位导致LiFSI电池达到更低的截止电压,因此,锂电镀比KFSI电池更快,限制了容量。从图6a,b中降低的K+电解质浓度梯度来看,靠近集流体的石墨电极背面电解质K+浓度明显高于Li+浓度,表明石墨可插入的K+更多,从而获得更大的容量。即使在4C的高充电速率下,KFSI电池也可以获得19%的容量,而LiFSI电池只有1%。
总结和展望
本工作充分表征了K离子电解质体系的离子传输和热力学性质,并将它们与锂离子体系进行了比较。此外,还开发了一种K金属制备方法,使电解质表征具有足够的稳定性。结果表明:在浓度低于2M时,KFSI:DME电解质的盐扩散系数和阳离子转移数均显著高于LiFSI电解质。较高的盐扩散系数和阳离子转移数减少了离子浓度梯度的形成和浓差过电位,从而证实了KIB具有提高倍率性能和低温性能的潜力。它们的离子电导率在20°C时相似,LiFSI略高,直到~1.7M,可能是由于KFSI盐解离不足导致。热力学因子随浓度变化的行为表明,与Li+相比,K+的溶剂和离子相互作用较弱。本工作还对K离子和Li离子金属||石墨电池进行DFN模拟,证明了KFSI:DME电解质具有更快的传输特性,从而提高了充电速率。总的来说,本研究证明,增加阳离子尺寸和降低K+电荷密度,从而减弱溶剂-离子和离子-离子相互作用,有利于实现大功率电化学储能系统。
审核编辑:刘清
-
锂离子电池
+关注
关注
85文章
3260浏览量
77999 -
锂电池
+关注
关注
260文章
8200浏览量
172161 -
电解质
+关注
关注
6文章
821浏览量
20190 -
OCV
+关注
关注
0文章
25浏览量
12579 -
XPS
+关注
关注
0文章
97浏览量
12066
原文标题:牛津大学最新Nature子刊:钾电与锂电电解质离子输运及热力学性质差异的根源!
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
清华大学:自由空间对硫化物固态电解质表面及内部裂纹处锂沉积行为的影响
陈军院士团队最新Angew,聚合物电解质新突破

镁合金牺牲阳极与电解质接触不良的原因

Li3MX6全固态锂离子电池固体电解质材料
一种薄型层状固态电解质的设计策略
无极电容器有电解质吗,无极电容器电解质怎么测
聚合物电池和三元锂电池的区别
具有密集交联结构的明胶基水凝胶电解质(ODGelMA)
如何判断电解池的电势高低?
电解质电极信号采集控制板

请问聚合物电解质是如何进行离子传导的呢?





 工商网监
工商网监
评论