 认识石榴石固态电解质的表面再生和反应性
认识石榴石固态电解质的表面再生和反应性
研究背景
基于固体电解质(SE)的锂金属电池可以实现高能量存储设备,因为它们与锂金属阳极和高压阴极具有潜在的兼容性。它们比目前最先进的液体锂离子电池电解质具有更高的热稳定性。在迄今为止探索的各种SE中,掺杂了LLZO((Li7La3Zr2O12)) 柘榴石具有 0.1–1 mS cm-1的高室温 (RT) 离子电导率以及相对广泛的电化学稳定性,这使它们成为商业应用的有希望的候选者。
众所周知,LLZO与大气中的微量水分和二氧化碳发生反应。这导致晶格中的Li与H交换形成质子化的LLZO(H++xLi7–xLa3Zr2O12)以及氢氧化锂和碳酸盐表面层。质子化导致晶格收缩和对称性从Iad 到I3d的变化,由此产生的异质表面层具有非常低的锂离子电导率,因此当LLZO与锂金属阳极配对时,增加了界面电阻。这导致Li-LLZO-Li金属界面处的电流分布不均匀,从而降低了临界电流密度(ICCD),其中锂金属枝晶成核并使电池短路。
已经报道的LLZO再生的不同方案中样品已在各种气体和温度下进行处理。尽管如此,再生表面所需的最低温度尚未明确确定,并且尚未系统研究不同气体的影响。另据报道,LLZO的过度加热会导致焦绿石的形成。已经使用体X射线衍射(XRD)研究了这种不可逆的分解,并且已经报道了一系列起始温度。由于分解始于LLZO的表面,因此需要使用表面敏感技术进行仔细研究,以准确确定该反应的起始温度。最后,即使已经表征了表面层的组成,但对导致表面层形成的起始温度和反应机理知之甚少;例如,关于LLZO与CO2的直接反应性存在相互矛盾的报告。LLZO表面的极端敏感不仅需要表面灵敏度高的表征技术,还需要原位技术来了解LLZO的再生和反应性。
成果简介
近日,剑桥大学Clare P.Grey利用近环境压X射线光电子能谱和掠入射X射线衍射两种表面敏感技术研究了不同气体环境下表层的分解。石榴石的清洁表面分别在 400°C和 500 °C 以下直接与水分和二氧化碳发生反应。这表明额外的CO2处理石榴石需要浓度控制。通过在O2下加热以及避免 H2O 和 CO2,在不使用任何夹层的情况下制备的对称电池界面电阻小于 10 Ω cm2;沉积电流 >1 mA cm–2且没有枝晶萌生。
该工作以“Understanding the Surface Regeneration and Reactivity of Garnet Solid-State Electrolytes”为题发表在ACS Energy Letters上。
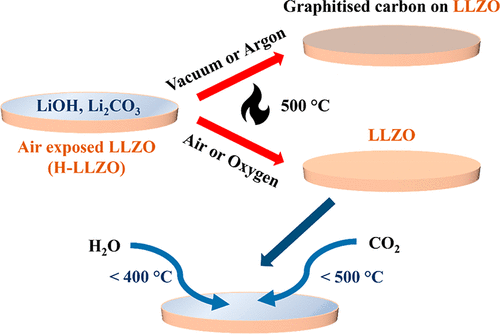
Scheme 1 不同气氛及温度条件对生产清洁LLZO表面的影响
研究亮点
(1) 利用近环境压X射线光电子能谱和掠入射X射线衍射两种表面敏感技术研究了不同气体环境下LLZO表层的分解。
(2) 研究发现,在氧气气氛(1 mbar及以上)下加热到500°C会导致干净的石榴石表面,而低氧分压(即氩气或真空)会导致额外的石墨碳沉积。
(3)在不使用任何夹层的情况下制备的对称电池界面电阻小于 10 Ω cm2;沉积电流 >1 mA cm–2且没有枝晶萌生。
图文导读
本研究将暴露在空气中的LLZO颗粒在不同的气体环境(真空、氩气、静态空气和流动空气)下加热,研究其再生过程,并采集原位掠入射X射线衍射(GIXRD)图来捕捉表面结构的变化。在真空、氩气、干燥空气和氧气加热下,采集了原位近常压X射线光电子能谱(NAP-XPS)光谱,绘制了表面的化学组成图。在氧气下,形成了完全再生和清洁的LLZO表面。然后在氧气下加热再生样品,并在H2O蒸气、CO2和H2O蒸气+CO2混合物下冷却样品,收集原位NAP-XPS光谱,以了解LLZO表面层的形成。最后,通过在氧气中加热空气暴露的LLZO样品,并在冷却过程中避免CO2和H2O,获得了较低的Li-LLZO界面电阻(<10ω cm2),在1mAcm-2以上的电流下无枝晶生长。
不同气体环境下的GIXRD测量:
Al-LLZO(由Al0.36Li5.92La3Zr2O12组成)粉末是用固态法合成的,热压,然后切成球团(相对密度约为99%)。通过同步加速器XRD确定了球团的相纯度。然后在不同气体环境下进行了GIXRD测试。由于设置的限制,不能在纯氧下加热,但测试了真空(0.01 mbar)、氩气、静态空气和流动空气(1atm)环境。所有样品首先在RT下暴露于空气中20分钟,然后放入光束线上的GIXRD装置中。掠入射角为0.1°,相当于探测深度约为3 nm。(LLZO)样品在控制的气体环境中以100°C的步骤加热到800°C,收集每个温度下的GIXRD图像然后冷却到室温。在RT下,所有暴露在空气中的样品都观察到了碳酸锂和氢氧化锂(图1)。在真空加热下,LLZO颗粒上的Li2CO3和LiOH在500℃以上(分别为▼和●的反射,图1,顶部)分解,分别在700℃和800℃时几乎完全消失。这与以前对真空下纯Li2CO3分解的观察结果一致。在800℃时,观察到Li2ZrO3和LaAlO3的形成(⧫和*标记的反射,图1,顶部),但没有观察到焦绿石La2Zr2O7。
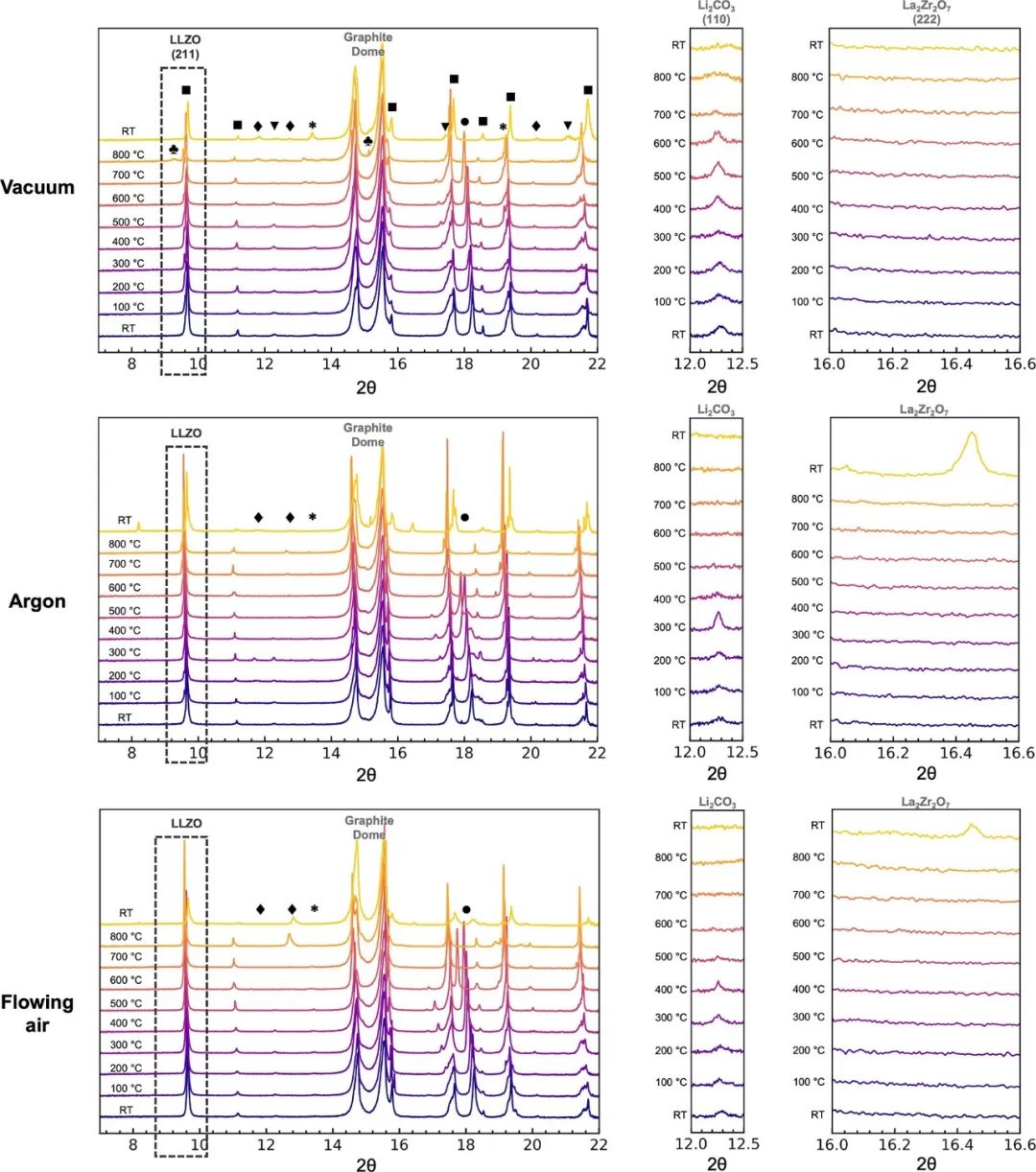
图1在不同的气体环境下,从室温到800°C,以100°C的增量加热,然后冷却到室温,暴露在空气中的样品的GIXRD(λ = 0.8856 Å)图形。第二和第三列图像是分别对应于Li2CO3(110)和La2Zr2O7(222)反射的区域的放大版本。黑色虚线框和■代表LLZO反射,代表⧫Li2ZrO3,*代表LaAlO3,●代表LiOH,▼代表Li2CO3。在800°C的真空情况下,用★表示的峰不能被指标化为任何已知的化合物,这些峰在冷却到RT时消失。随着样品的加热,LLZO(211)峰向低2θ方向移动是由于热晶格膨胀;在800°C和RT下,由于快速的晶格收缩,所有相的反射都出现了明显的不连续性。
在氩气加热下,Li2CO3和LiOH分别在400℃和500℃的较低温度下发生分解。加热到800°C并没有导致LLZO分解,但在样品冷却到RT后检测到焦绿石La2Zr2O7,表明LLZO在氩气下高温分解。在流动空气下,Li2CO3和LiOH分别在400℃和600℃以上发生分解,样品在氩气下冷却到RT后,再次观察到La2Zr2O7焦绿石的分解,表明在流动空气下也发生了类似的分解机理。有趣的是,当样品在静态空气中加热(类似于箱形炉的条件下),Li2CO3和LiOH仅在600℃以上才会分解,与真空情况相同。在静态空气中加热至500°C以上,LLZO的大量分解和焦绿石的形成也被观察到,而在600°C以上,LLZO信号完全消失,这表明气体环境中的微量水分没有被气体流动或抽真空去除,导致LLZO更快地分解。我们注意到,Li2O的蒸发被水的存在显著地增强(形成挥发性更强的产物LiOH), GIXRD在静态空气中加热时看到的许多降解产物。
不同气体环境下的NAP-XPS测量:
利用NAP-XPS对GIXRD观测结果进行了补充,并绘制了LLZO球团加热过程中表面化学成分的演变图。这些颗粒被抛光并储存在一个手套盒中,然后暴露在空气中20分钟,然后被泵入光束线上的NAP-XPS仪器。入射能量的调整方式是探测到的抛射光电子的动能为~ 200 eV;这相当于所有XPS测量的探测深度约为3 nm。样品在受控气体(1 mbar,真空:2 × 10−8 mbar)环境中加热至500°C,温度为100°C,然后冷却到RT,在这些过程中收集XPS光谱。C 1s、O 1s和La 3d的三维光谱演化如图2所示,。所有样品的C 1s XPS光谱在RT处显示两个信号(图2)。由于碳酸锂预计会在暴露在空气中的样品表面形成,高eV处的峰与289.9 eV处的Li2CO=峰一致,其他元素的XPS光谱也发生了相同幅度的偏移。在所有样品中,尤其是在低温下,均观察到明显的峰宽、峰移和强度变化,使分析变得复杂;因此,图2中也比较了500°C数据,因为它们最容易分析。
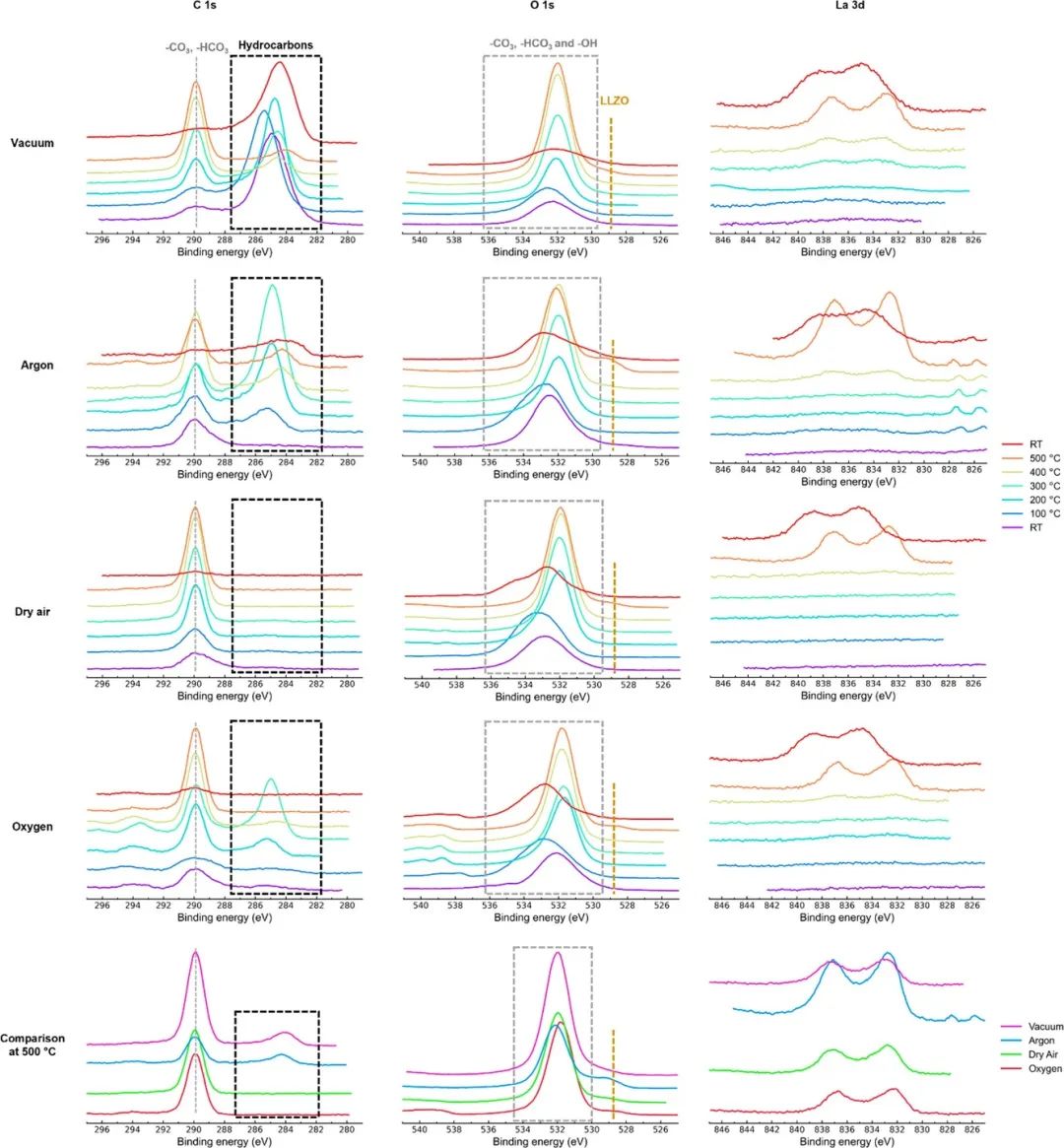
图2 暴露在空气中的样品的C 1s、O 1s和La 3d XPS光谱,在室温至500 °C的不同气体环境下以100 °C的增量加热,然后冷却至室温。灰色虚线和灰色虚线框代表Li2CO3,金色虚线代表LLZO,黑色虚线框代表表面吸附的碳氢化合物和石墨化碳。最后一行比较了不同气体环境下500°C下的C 1s、O 3s和La 3d XPS光谱。
当样品在所有环境中加热时,在C 1s光谱中都观察到一个~ 285 eV的额外峰(图2)。当样品在干燥空气和氧气下加热到500°C时,这个峰消失了。相比之下,这个峰在真空和氩气加热的样品中仍然存在,甚至在冷却到rt后仍然存在。这表明这个峰来自表面吸附(含sp3)的碳氢化合物,可能存在于真空室或手套箱中。在干燥的空气和氧气中氧化,但在低氧分压(真空和氩气)下石墨化,导致该峰向较低的eV (284.1 eV;见500°C下的对比,图2)。以前在真空下进行的原位XPS研究也进行了类似的观察。25,26我们的研究结果表明,在氩气加热下,LLZO表面也会产生石墨碳。当样品从RT加热到100℃时,宽大的O 1s峰向更高的eV移动。向更高eV的转变可以归因于表面-HCO3−物种由于表面解吸水而增强。在100℃以上,可以观察到峰值的锐化和向更高eV的转移。这种锐化可以归因于在高温下XPS测量和表面吸附水释放过程完成时更好的电荷补偿。现在看到的峰值可以被指定为一种CO3 2−物种。在所有气体环境中,O 1s XPS光谱在500°C约为529 eV时出现了一个额外的小峰(图2)。
在500°C时,La 3d和Zr 3d光谱中也出现了类似的尖锐峰,在RT时看不到(图2)。因此,O 1s谱中529 eV处的峰被分配到LLZO晶格中,证实了500°C时LLZO的再生(尽管500°C时C 1s Li2CO3峰的存在表明表面层的不完全分解)。在真空、氩气和氧气条件下冷却到RT后,LLZO O 1s峰值仍然存在,但在干燥空气条件下则不存在。GIXRD观察表明,如果样品在氩气或流动空气(以及氧气)下处理,加热至500°C可以分解LLZO的表层。虽然XPS光谱显示500°C时表层的不完全分解,但这种差异可能是由于在两种装置中测量样品温度的方式不同。当样品在真空和氩气下加热时,观察到额外的石墨形成。虽然最近的研究表明,碳夹层降低了Li - LLZO界面电阻并改善了性能,由于在氩气和真空下加热,表面的石墨化不需要是均匀的,而且当LLZO用于SE对Li金属时,任何不均一性都可能导致电流聚焦。最后,避免温度超过500°C将防止焦绿石的形成,无论样品在何种环境下加热以再生LLZO。
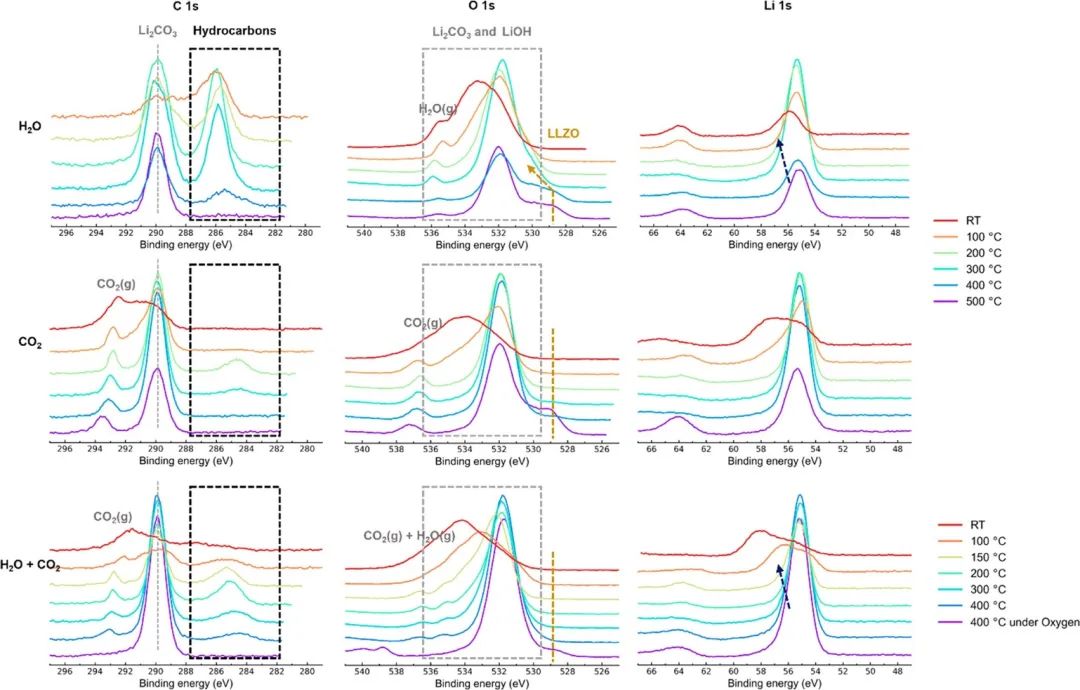
图3。在不同气体环境下,LLZO从500°C冷却到RT冷却过程中C 1s和O 1s的XPS光谱。灰色虚线代表Li2CO3,金色虚线代表LLZO,黑色虚线框代表地表附加的碳氢化合物相关物种,深蓝色虚线代表LiOH。
利用NAP-XPS测量研究表层形成的起始温度:
为了防止在冷却过程中发生表面层的改变,确定LLZO与水分和CO2反应的起始温度是很重要的。LLZO样品首先在500°C的氧气下处理1小时,以允许表面再生,LLZO晶格峰在~ 529 eV的O 1s光谱中被观察到,证实大部分表面污染物被去除。接下来,通过手动打开阀门,将H2O蒸汽引入反应室,阀门连接到一个含水的石英管。然后采集500℃冷却至室温下的XPS光谱。LLZO O 1s的晶格峰在引入水蒸气时仍然存在,而C 1s的光谱显示出一个单峰,这是由上面讨论的残余Li2CO3造成的。当样品冷却到400°C时,随着Li2CO3峰强度的持续降低,在C 1s谱中出现了约286 eV的额外峰。在O 1s谱中,Li2CO3峰变宽,而LLZO晶格峰保持不变。额外的峰归因于氧化表面吸附的碳氢化合物(位移对应于含醚的碳氢化合物),正如文献中所观察到的,当水被引入XPS室时。在300℃时,O 1s谱中的LLZO峰移向更高的eV,表明LLZO发生质子化。Li 1s峰变宽,这与LiOH的形成有关。C 1s谱中附加的(~ 286 eV)峰强度增加,现在可与Li2CO3峰相媲美。当样品冷却到200℃以下时,LLZO晶格峰完全消失,表明LLZO表面广泛形成LiOH,这与最近的一项研究一致:
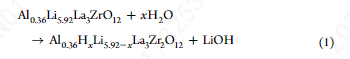
C 1s谱上的附加峰在冷却到RT后仍然存在。
即使在冷却到RT后,C 1s光谱中的附加峰仍然存在。为了探测二氧化碳与LLZO的反应,样品在O2下加热,CO2被引入反应室。当样品冷却到RT时,采集XPS光谱。在500℃时,在O 1s区~ 529 eV处观察到LLZO晶格峰,这是CO2引入时留下的峰。碳原子1s谱为单峰,对应于Li2CO3。当冷却到400℃时,LLZO的峰强度明显下降,表明LLZO与CO2发生了直接反应。这可以表示为
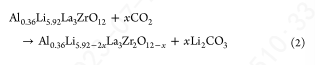
这种形式的反应需要氧阴离子的萃取,形式上是通过Li2O的萃取。因此,这个反应很可能只局限在表面,因为这个反应需要在晶格中产生大量的氧空位才能进入样品的主体。与CO2的反应为酸碱反应,母氧化物相Li2O的碱度(和Li+迁移率)促使反应生成Li2CO3。而母氧化物La2O3较碱性,形成La2(CO3)3,在高温下通过形成La2O2CO3分解;在1atm的CO2.38下,La2O2CO3要到950℃才会分解形成La2O3。因此,以下形式的反应也可能发生,这里写的是父LLZO相,以说明一个可能的分解途径:
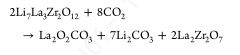
虽然这个反应可能只发生在表面,因为它涉及La3+和Zr4+的迁移,它不需要在LLZO中形成氧空位。这也代表了高温下焦绿石相形成的一种合理机制,其中可以发生La3+/Zr4+迁移。此外,当在静态空气(或任何封闭容器)中加热时,Li2CO3在较低温度下分解释放出的任何二氧化碳,都可以反应生成氧化型碳酸镧(La2O2CO3)。任何微量水的存在也会导致不需要产生多重氧空位的反应:
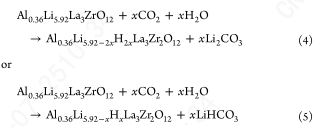
当样品冷却到300℃以下时,LLZO晶格峰完全消失。在C 1s谱中,在~ 284.8 eV附近发现了一个新的峰,这可能是表面吸附的烃类。当样品冷却到100℃时,这个额外的峰在C 1s光谱中完全消失,Li2CO3峰明显展宽。Li2CO3 O 1s峰展宽,表明LLZO与CO2发生了广泛的反应,或者样品发生了剧烈的充电。这些结果也解释了在干燥空气下加热再生LLZO后,在RT下观察到的O 1s光谱的增宽;干燥空气中微量的CO2会在400℃以下与LLZO发生反应,并使表面钝化。在环境大气中,H2O (~ 30mbar)和CO2 (~ 0.42 mbar)的含量都是微量的。在这种情况下,为了检查LLZO的反应性,LLZO首先在O2下热处理,然后在H2O和CO2的1:1混合物中冷却到RT。在400°C时,LLZO峰减少并向更高的eV移动,表明发生了质子化。在C 1s光谱中观察到一个额外的峰,就像在H2O和CO2的情况下。
在进一步冷却后,Li 1s、C 1s和O 1s的峰值演化基本上是纯H2O和纯CO2情况下的结合,这表明正在发生方程式3、4和5所描述的反应。这些结果表明,在进行LLZO再生的环境中,CO2水平需要与H2O水平相控制。这对处理LLZO提出了一个独特的挑战,因为二氧化碳水平通常不被监测和控制。我们的结果应该与最近报道的LLZO/薄膜阴极模型系统(以LiNi0.6Mn0.2Co0.2O2或LiCoO2为阴极)在CO2存在的情况下烧结,导致阴极和LLZO的分解增强;这些结果突出了碳酸盐形成的强烈驱动力量。即使LLZO表面通过LLZO -阴极界面的形成得到保护。相比之下,在纯氧或惰性气氛(N2)下加热到700°C时,LLZO -阴极界面电阻较低。在500°C的加湿氧气中,界面电阻较高,但在700°C的加湿氧气中,界面电阻下降到与纯氧结果相当的值。相比之下,在当前的研究中,发现任何水分都会导致500℃以上的LLZO广泛分解;在之前的阴极- LLZO系统中,在水分存在的情况下,LiOH没有降解,这归因于阴极膜对LLZO表面的保护,这有助于减少LiOH在潮湿环境下的蒸发。在我们对裸球团的研究中,惰性气氛(氩)导致了LLZO表面的石墨沉积,并且发现只有氧气是再生LLZO的理想气体。
电化学研究:
由于NAP-XPS和GIXRD测量结果表明,氧气加热(分压1 mbar及以上)会导致表层分解,并可导致LLZO完全清洁的再生,因此LLZO颗粒在氧气下处理,然后转移到手套箱中没有暴露在空气中,通过自定义设置,用氩气吹扫。组装Li - LLZO - Li对称电池,进行阻抗测量(图4),使用等效电路模型拟合数据。Li - LLZO的界面电阻小于10 Ω cm2,而手套盒内刚打磨过的样品的界面电阻为>500 Ω cm2。为了在锂枝晶形成之前估计ICCD,我们将单向电流施加到对称电池中。ICCD被发现为>1 mA cm−2,与报道的具有界面电阻>10 Ω cm2, ICCD <0.5 mA cm−2相比,这表明通过优化方案来降低界面电阻,LLZO石榴石中可以实现高电沉积电流密度。
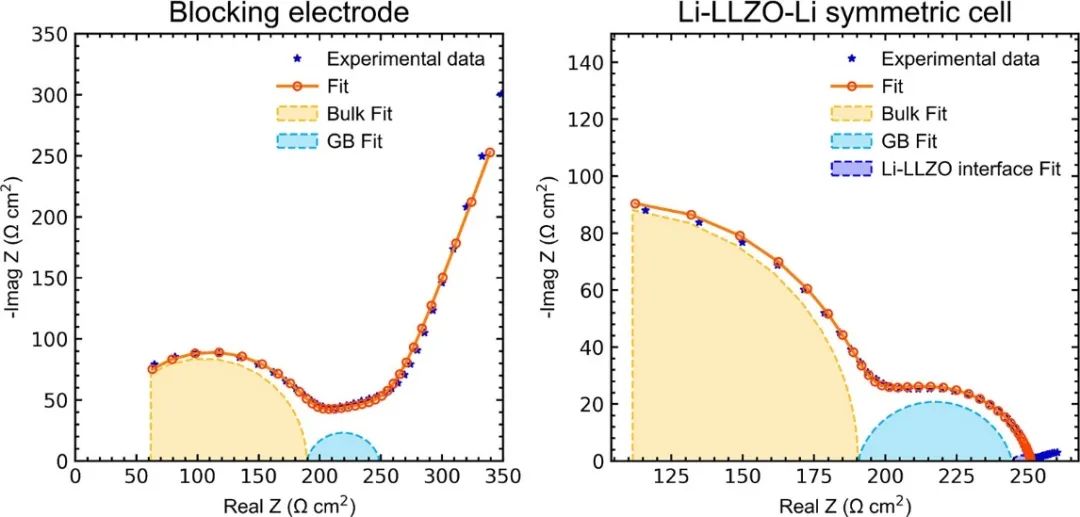
图4。左)LLZO颗粒在阻塞条件下的阻抗谱,拟合等效电路模型来确定体相和晶界贡献;右)Li-LLZO-Li对称电池,显示体相、晶界和Li-LLZO界面
总结与展望
综上所述,本工作表明,无论气体环境如何,石榴石暴露在空气中形成的表面层(LiOH和Li2CO3)都可以通过加热至500°C进行分解。此外,在真空和氩气条件下,LLZO的加热过程中还生成了石墨碳。虽然氩气是LLZO再生最常见的环境,但作为SE时,非均相石墨化会导致电流聚焦,影响LLZO的性能。在氩气和流动空气中加热至600 ~ 700℃时,LLZO会分解成辉绿石,而在真空中则不会。清洁的LLZO表面在400℃以下与水分发生反应,在500℃以下与CO2发生反应的直接证据显示。提出了一种优化的LLZO再生方案,结果表明,在不使用Li金属与LLZO之间的任何中间层的情况下,LLZO颗粒的界面电阻可达到10 Ω·cm2以下。这些结果意味着,除了用于组装液电解质锂离子电池的干燥室外,LLZO的处理还需要更专门的二氧化碳水平控制。
审核编辑:刘清
-
锂离子电池
+关注
关注
85文章
3264浏览量
78142 -
电阻器
+关注
关注
21文章
3828浏览量
62571 -
加速器
+关注
关注
2文章
813浏览量
38340 -
XRD
+关注
关注
0文章
133浏览量
9292 -
固态电解质
+关注
关注
0文章
86浏览量
5503
原文标题:剑桥大学Clare P.Grey−ACS Energy Letters:认识石榴石固态电解质的表面再生和反应性
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
超声波焊接有利于解决固态电池的枝晶问题
清华大学:自由空间对硫化物固态电解质表面及内部裂纹处锂沉积行为的影响
陈军院士团队最新Angew,聚合物电解质新突破

镁合金牺牲阳极与电解质接触不良的原因

一种薄型层状固态电解质的设计策略
半互穿网络电解质用于高电压锂金属电池
固态电池中复合锂阳极上固体电解质界面的调控

固态电池的优缺点 固态电池与锂电池比较
无极电容器有电解质吗,无极电容器电解质怎么测
请问聚合物电解质是如何进行离子传导的呢?





 工商网监
工商网监
评论