 基于cAMOEBA极化力场的燃烧气相分子的输运参数
基于cAMOEBA极化力场的燃烧气相分子的输运参数
01引言
近几十年来,燃烧化学得到了长足的发展[Prog. Energy Combust. 2014, 43.]。应用了许多实验和理论方法来研究燃烧相关问题,包括相关化学物质的分子间势能、传输特性和碰撞能量转移计算[Prog. Energy Combust. 2021, 83.]。然而,由于燃烧反应过程被认为是一个复杂的"网络结构",涉及许多反应和大量中间产物, 很难通过实验确定不同化学物质的输运参数[Combust. Flame. 2014, 161.]。因此,理论计算就显得尤为重要。因此,建立精确的燃烧动力学模型是燃烧化学领域最重要的任务之一[Combust. Inst. 2021, 38.]。
02成果简介
在本研究中,我们报告了一个针对燃烧相关分子的简化但精确的通用AMOEBA极化力场,称为Combustion-AMOEBA或cAMOEBA。其中,气体分子弛豫优化是通过鸿之微DS-PAW软件进行计算(其为cAMOEBA极化力场的构建而服务)。通过消除永久原子偶极子和四极子,保留明显的极化,并定义每个分子种类(包括烷、烯、 炔、醇、过氧化物和醛)的原子类型,构建了简化的通用cAMOEBA力场,并利用在QCISD(T)/CBS理论上获得的基准结果进行了验证。这样就避免了在原始AMOEBA(Poltype/MP2)力场中对每个新分子的永久原子多极进行参数化的繁琐步骤,从而能够以较低的计算成本对大量分子进行精确的高通量计算。
此外,我们使用cAMOEBA和AMOEBA (Poltype/MP2)计算的约100种不同分子和四种浴气(He、Ne、Ar和N2)的输运参数σ和ε的平均差异分别为0.09%和1.27%,这表明通用cAMOEBA力场与原始AMOEBA(Poltype/MP2)力场具有良好的一致性,在原始力场中,每个小分子的多极力场参数都是通过量子力学计算获得的。我们的结果还表明,在从一种浴气中获得纯气体分子Lennard-Jones参数时,Lorentz-Berthelot组合规则比Waldman-Hagler规则更适用, 而Waldman-Hagler组合规则则更适合从所有四种浴气中获得这些参数。则更适用于从所有四种浴气中获取这些参数。使用cAMOEBA获得的纯气体参数可用于为燃烧建模开发高质量的传输特性数据库。
03图文导读
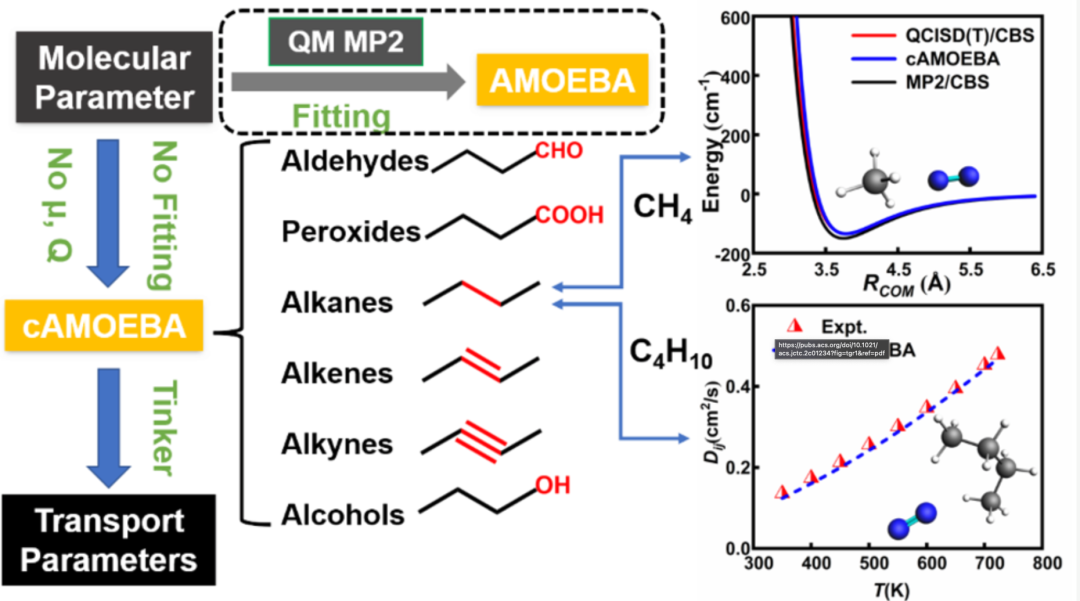
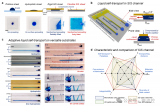
图1:对目标分子进行结构优化,为燃烧分子构建简化且高精度的极化力场
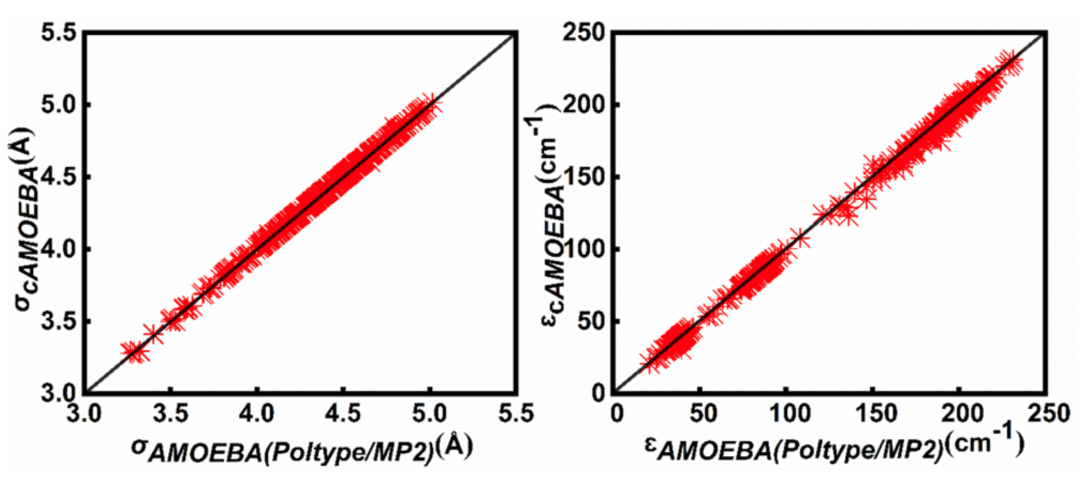
图2:基于cAMOEBA力场计算的分子在不同浴气下的输运参数
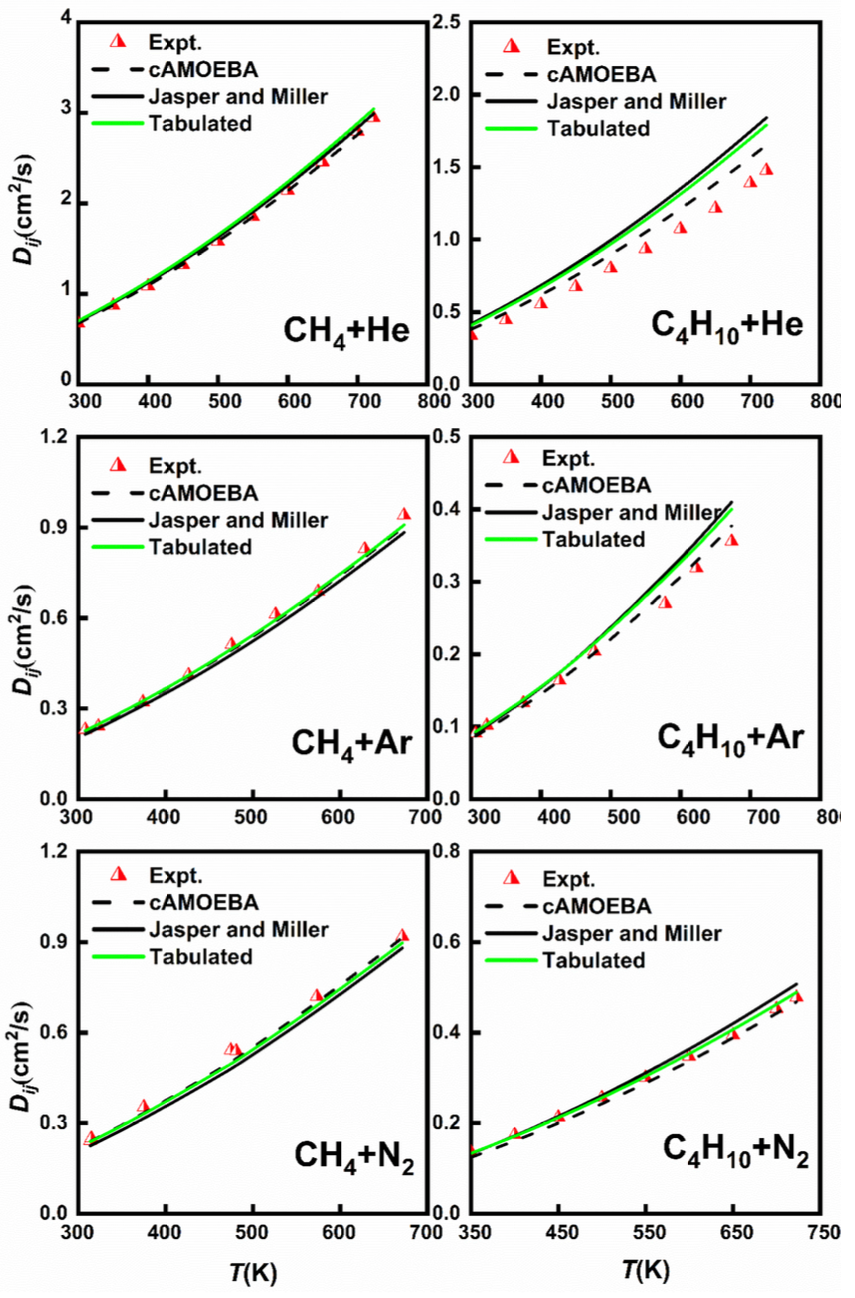
图3:通过不同的力场(Jasper and Miller, Tabulated values和cAMOEBA),对比选择分子的二元扩散系数
04小结
在本研究中,我们报告了用于燃烧相关分子建模的cAMOEBA力场的构建情况。该力场的目的是为更广泛的化学空间提供一个通用的、随时可用的力场, 而无需繁琐的参数化程序。目前的参数化已经涵盖了烷、烯、炔、醇、过氧化物和醛。通过将分子间势能与高水平的QM计算进行比较,以及将预测的传输特性与实验值进行比较,对力场进行了验证。
值得注意的是,与AMOEBA力场一样,cAMOEBA力场也是基于通用力场构建方案获得的力场,可提供精确的分子间相互作用。因此,cAMOEBA力场不仅可用于推导气体的L-J参数,还可用于其他通用模拟,如分子动力学模拟和相关的基于动力学的采样技术。使用新力场生成L-J参数的适用性取决于估算方法。洛伦兹-贝特洛组合规则更适合于选择一种浴气来计算纯气体的L-J参数。用该L-J参数计算的理论粘度与实验结果之间的误差小于7%。实验结果的误差小于7%。Waldman-Hagler组合规则适用于组合四种浴气来获得纯气体的L-J参数。用这种方法计算分子理论粘度得到的L-J参数分子的L-J参数与实验值非常接近,误差小于9%。最后,基于我们的力场和ODDM方法,我们还计算了约100种燃烧物的L-J参数,用于计算燃烧过程中的输运特性。
审核编辑:刘清
-
计算机
+关注
关注
19文章
7490浏览量
87894
原文标题:文献赏析|基于cAMOEBA极化力场的燃烧气相分子的输运参数(马建毅)
文章出处:【微信号:hzwtech,微信公众号:鸿之微】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
气敏膜传感器工作原理是什么
气敏元件工作时加热的目的是什么
气敏电阻为什么要加热使用
电力场效应管的结构和特性
电力场效应管的保护措施
电力场效应管的安全工作区
电力场效应管的动态特性和主要参数
电力场效应管的静态特性和主要参数
电力场效应管的主要类型和工作原理
控制阀气开气关的选择原则
极化继电器的工作原理是什么
极化继电器衔铁的偏转方向取决于
极化继电器的工作原理是什么 极化继电器工作原理图
利用柔性超亲水夹层通道,实现液体在任意界面上的自发定向输运





 工商网监
工商网监
评论