 上海交大JACS:单原子催化,非晶态载体更具优势!
上海交大JACS:单原子催化,非晶态载体更具优势!
通讯作者:占光明、么艳彩、张礼知
通讯单位:上海交通大学
文章链接:https://pubs.acs.org/doi/10.1021/jacs.3c13834
01
导读
氯气(Cl2)在有机合成、家用漂白剂和水处理中有着广泛的应用。工业生产Cl2需要在饱和氯化钠以及强酸性溶液条件下(pH值约2)下进行电解,导致阳极氯析出反应(CER)需要使用到大量DSA(负载于Ti基底的Ru/Ir氧化物)来加速电化学反应。为了可持续发展,减少贵金属用量和提高性能是CER电极发展的迫切要求。
02
成果简介
作者报道了非晶态钛氧化物载体能够有效调节Ir单原子的配位环境,从而实现钛阳极高效持久的析氯反应。实验和理论结果表明,在非晶钛氧化物和结晶钛氧化物上分别形成了四配位Ir1O4和六配位Ir1O4结构。有趣的是Ir1O4位点表现出优越的CER性能,其质量活性分别是IIr1O4和DSA的10倍和500倍。此外,Ir1O4阳极表现出优异的200小时耐久性,远远超过Ir1O4阳极(2小时)。机理研究表明,Ir1O4中的不饱和Ir位点是CER的活性中心。Ir1O4的无定形结构和受限的水解离协同阻止了O在Ti基底上的渗透,有助于其长期的CER稳定性。该成果以“Engineering the Coordination Environment of Ir Single Atoms with Surface Titanium Oxide Amorphization for Superior Chlorine Evolution Reaction”为题发表在国际顶级期刊Journal of the American Chemical Society上。
03
研究亮点
1,作者提出了钛氧化物载体的结晶性能够调节单原子Ir的配位结构:在非晶钛氧化物和结晶钛氧化物上分别形成了四配位Ir1O4和六配位Ir1O6结构。单原子Ir的结构差异将影响物种的吸附。
2,Ir1O4显示出优异的CER性能,过电位仅为71.8 mV@10 mA cm-2,且具有200小时耐久性。
3,实验与模拟结果显示,氧化层的非晶化结构以及缓慢的水解离有效阻止了氧渗透到Ti基底内部,提高了Ti的抗钝化能力。
04
研究内容
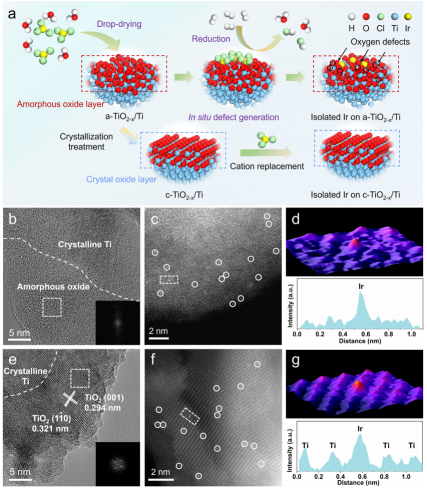
图1 合成及电镜表征:(a)在a-TiO2-x/Ti和c-TiO2-x/Ti上合成单原子Ir的示意图;(b,e)a-TiO2-x/Ti和c-TiO2-x/Ti的HRTEM图像;(c,f)a-TiO2-x/Ti和c-TiO2-x/Ti的AC HAADF-STEM图像;(d,g)分别沿着(c)和(f)中矩形标记的区域对应的三维AOGF映射和线强度分析。
通常,天然形成的钛氧化物的结晶度较差,其内部有晶态Ti,表面有非晶态钛氧化物层,边界清晰(记为a-TiO2-x/Ti)。通过对a-TiO2-x/Ti进行高温处理和慢冷相结合的结晶处理,制备出相应的晶体(c-TiO2-x/Ti),使其逐渐由非晶态转变为晶态。
接着,作者利用缺陷工程策略和金属-载体相互作用,将单原子Ir固定在不同晶相的钛氧化物上。通过HAADF-STEM可以看到,Ir分别分散在a-TiO2-x/Ti(图1b、c)和c-TiO2-x/Ti表面(图1e、f)。经三维原子重叠高斯函数拟合(3D AOGF),如图1d、g所示,其显示Ir呈现高度分散的孤立结构,与线强度分析一致。
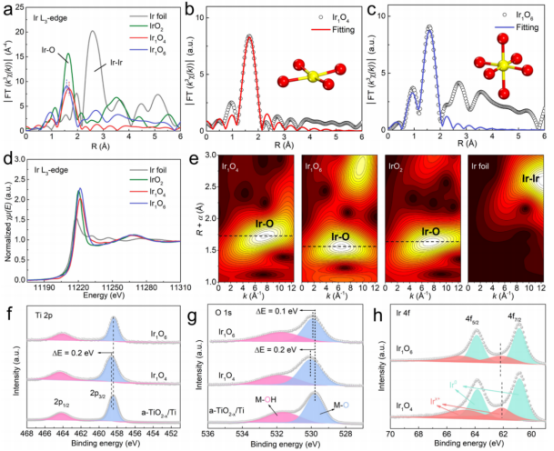
图2 结构表征:(a)Ir的L3边缘FT-EXAFS谱图;(b,c)Ir的L3边缘FT-EXAFS拟合曲线;(d)Ir的L3边缘XANES谱图;(e)小波变换光谱;(f-h)Ti 2p、O 1s、Ir 4f的XPS谱图。
接着,利用XAFS和XPS检测了单原子Ir的局部配位环境和电子态。两个样品的FT-EXAFS谱图均显示一个主峰,对应Ir-O配位,而没有出现Ir-Ir配位,表明Ir呈原子分散结构(图2a)。通过拟合EXAFS光谱(图2b、c),结果表明,在a-TiO2-x/Ti上,Ir仅与4个相邻的O配位(记为Ir1O4),表明Ir具有高度配位不饱和的特征;而负载于c-TiO2-x/Ti上的Ti与6个相邻的O配位(记Ir1O6。因此,Ir1O4中的Ir-O散射路径(~1.67 Å)比Ir1O6的Ir-O散射路径(~1.58 Å)更大,这是由于在非晶基底上,Ir-O原子间距被拉伸所致;图2d的小波变换也证实了这一结果(Ir1O4具有较高的R和k值)。
图2e的Ir的L3边缘XANES谱图显示,与Ir1O6相比,Ir1O4中的Ir的L3边缘的峰强度较低,即由载体非晶化引起Ir的平均价态较低。与a-TiO2-x/Ti基底相比,Ir1O4中Ti 2p3/2的峰偏移到更高的结合能处,表明在Ir沉积后,电子可能从Ti转移到单原子Ir。结合Ir1O4中明显正移的金属-氧峰(M-O)(图2g),可以推断电子转移发生在Ti-O-Ir上。相反,对于Ir1O6,Ti 2p3/2和M-O的峰位移较小,表明其Ti-O-Ir单元的电子转移较弱。这一推测也得到了Ir1O4中Ir 4f7/2具有较低的结合能的支持(图2h),表明电子从Ti基底转移到单原子Ir上。因此,Ti基底的表面氧化层的结晶度将强烈影响单原子Ir的局部配位环境与电子结构(配位饱和的Ir1-O6结构可能难以吸附Cl物种),从而提供明显不同的CER活性。
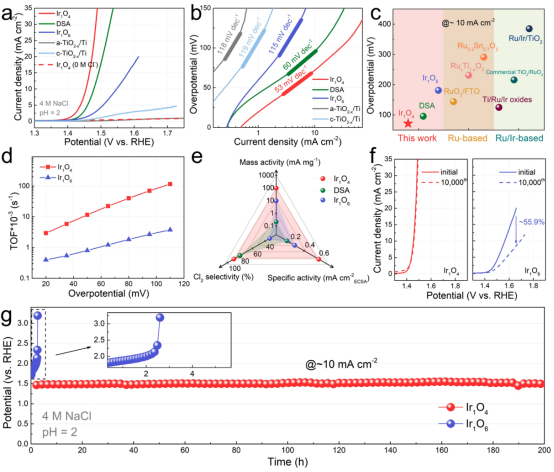
图3 CER性能:(a)CER极化曲线;(b)Tafel曲线;(c)不同催化剂的过电位比较;(d)Ir1O4和Ir1O6的TOF数值比较;(e)Ir1O4、Ir1O6和DSA的性能参数比较;(f)循环1万次后的极化曲线;(g)在10 mA cm-2下的计时电位曲线。
在pH=2的4 M NaCl中评估了Ir1O4、Ir1O6的CER性能,并将其与商业DSA、a-TiO2-x/Ti和c-TiO2-x/Ti进行了比较。图3a的LSV曲线显示,Ir1O4可以在更低的过电位下提供更高的电流密度。例如,在10 mA cm-2下,Ir1O4的过电位仅为71.8 mV,且Tafel斜率为53 mV dec-1,这一结果远优于Ir1O6(181.4 mV和115 mV dec-1), DSA (95.8 mV和60 mV dec-1),以及大多数的Ru和Ru/Ir基CER电催化剂(图3b、c)。
图3d进一步比较了Ir1O4、Ir1O6的转换频率(TOF)。例如,Ir1O4在过电位为110 mV时的TOF为0.12 s-1,比Ir1O6的TOF高31倍。同时,由ECSA归一化的比活性也揭示了Ir1O4具有更高的本征活性(图3e),优于商业DSA与Ir1O6。
在没有NaCl的情况下,Ir1O4几乎不产生法拉第电流(图3a),表明其OER活性极差。在电解质中加入NaCl后,Ir1O4的Cl2选择性达到90.0%,与Ir1O6(32.5%)和DSA(80.1%)形成鲜明对比(图3e),证实了其优异的Cl2选择性。更重要的是,Ir1O4表现出优异的耐久性,在循环1万次前后的极化曲线、以及200 h的长时间运行中都没有明显的活性衰减(图3f、g)。

图4 CER机制:(a)Ir1O4和Ir1O6中单原子Ir的局部配位构型;(b)差分电荷密度图;(c)CER过程的自由能图;(d,e)CER条件下的原位拉曼光谱;(f)吸附氯原子后,Ir1O4和Ir1O6的DOS分布;(g)反应后收集Ir1O4和Ir1O6,进行H2-TPR处理;(h)在m/z=36时,收集到的H2-TPR谱线。
作者采用DFT计算来理解Ir1O4优异的CER活性和活性位点。根据EXAFS拟合结果,构建了Ir1O4和Ir1O6的合理模型,如图4a所示。差分电荷密度图显示单原子Ir和基底之间存在电子相互作用,Ir1O4的Bader电荷转移(+1.15e)低于Ir1O6的Bader电荷转移(+1.40e),即更多的电子从a-TiO2-x/Ti转移到单原子Ir上。图4c显示了CER过程的自由能,Ir1O4上亲电不饱和Ir位点的Cl吸附能为-0.1 eV,比Ir1O6上配位O位点的Cl吸附能(+0.31 eV)更接近于0 eV,有利于Cl的吸附。
因此,暴露的单原子Ir是Ir1O4的Cl吸附位点,这也被原位拉曼光谱证实(图4d):随着反应电位的增加,在500 cm-1处出现了一个越来越强的Ir-Cl键拉曼信号。相比之下,在饱和配位的Ir1O6催化剂上,只在791 cm-1处观察到一个较弱的峰(O-Cl键)(图4e)。空间位阻效应增加了Cl在饱和Ir配位位点上的吸附难度,但有利于其在上配位O原子上的吸附。
通过态密度(DOS)进一步比较不同吸附位点与Cl之间的相互作用(图4f)。Ir1O4中Ir 5d和Cl 3p轨道的重叠比Ir1O6中O 2p和Cl 3p轨道的重叠要大,表明Cl与不饱和Ir位点的相互作用强于与饱和Ir的顶配位O的相互作用。通过H2程序升温还原(H2-TPR)进一步验证了Cl物种在Ir1O4和Ir1O6上的吸附强度,发现Ir1O4中Cl解吸峰的温度高于Ir1O6(图4g、h),证实了Ir1O4对Cl的吸附更强,这与理论计算结果和原位拉曼光谱中更强的吸附信号相吻合。上述分析可以简单理解:CER催化活性的巨大差异源于Ir1O6上Cl的吸附位点从顶部配位的O到Ir1O4中不饱和Ir位点的变化,从而影响了与Cl的结合强度。
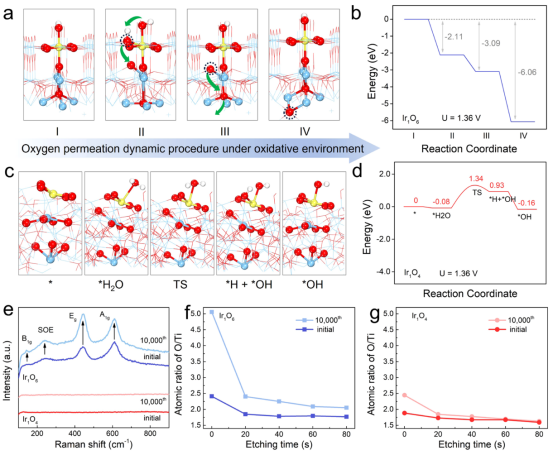
图5 失活机理研究:(a)氧化环境下Ir1O6可能的氧渗透动态过程;(b)氧渗透动力学过程的相应自由能图;(c)Ir1O4在水吸附和随后解离成羟基过程中的局部结构构型和过渡态;(d)水吸附和解离的自由能图;(e)Ir1O4和Ir1O6在10000圈循环前后的拉曼光谱;(f,g)不同蚀刻时间后O/Ti的原子比。
通过理论模拟分析了结晶钛氧化物的钝化过程(图5a、b)。由于整个CER过程发生在酸性溶液中,处于初始状态的Ir1O6表面以表面吸附羟基(*OHsurf)的形式暴露出来(阶段I)。在施加电位(1.36 V)下,表面配位饱和的Ir1O6会自发吸附另一个OH,吸附能为-2.11 eV,达到过饱和状态(阶段I~II)。初始态的*OHsurf由于位阻的排斥力进入亚表面,自发脱氢形成亚表面氧(Osub,阶段II~III),新的*OHsurf将占据原来*OHsurf的位点。在氧化层和金属层之间氧浓度差的驱动下,新形成的Osub可以沿着Ti-O-Ti自发地渗透到氧化层和金属层的界面中(阶段III~IV),从而使氧化层变厚,导致电子转移阻抗的急剧增加。总的来说,在Ir1O6上,OH的过饱和吸附(阶段I到II)、OH的脱氢渗透(阶段II到III)以及随后Osub的深度渗透(阶段III到IV)都是自发过程。
在氧渗透过程中,氧化层和金属层之间的氧浓度差是动力,而规则且排列紧密的Ti-O-Ti框架通过提供轴向通道促进了这一过程。对于Ir1O4,由于水解离存在较大的势垒(1.42 eV),阻止了额外的OH吸附(图5c、d)。因此,Ir1O4催化剂上的氧渗透和Ti基底的钝化被抑制。
反应前后电极的拉曼光谱证实了Ir1O6上氧化层的增厚(图5e),反应后在143、239、443和610 cm-1处的拉曼信号明显增加,对应金红石型TiO2结构,而Ir1O4没有观察到这种情况,这与其无定形氧化层的无序特性有关。
通过Ar离子蚀刻的XPS光谱进一步验证了这种抗钝化机制,定性分析了电极表面和亚表面上O/Ti原子的比例(图5f、g)。新鲜的Ir1O4和Ir1O6电极表面的O/Ti原子比很低,小于2。然而,反应后Ir1O6的表面O的比例明显增加,这是由于氧的吸附能力过大。随着刻蚀时间的增加,O/Ti比值甚至高于初始样品(>2),证实了氧渗透到电极中,使电极钝化(图5f)。经过1万次循环反应后,Ir1O4的表面O比例仅略有增加,而亚表层的O/Ti比例与初始样品非常接近,有力地验证了氧化层非晶化的抗钝化能力(图5g)。上述分析可以简单理解:对于Ir1O4,无定形氧化层结构和较高的水解离势垒,有效阻止了氧渗透到Ti基底内部,抑制了Ti钝化,使Ir1O4显示出优异的CER稳定性。
05
总结与展望
综上所述,本文证明了表面钛氧化物非晶化是一种有效的策略,可用于调节单原子Ir的配位环境,以实现高效和稳定的CER。所制备的Ir1O4电极在10 mA cm-2下过电位仅为71.8 mV,质量活性为95 mA mgIr-1,远远优于Ir1O6和DSA。原位拉曼光谱和理论模拟表明,CER催化活性的巨大差异源于Ir1O6上Cl的吸附位点从顶部配位的O到Ir1O4中不饱和Ir位点的变化,从而影响了与Cl的结合强度。更重要的是,钛氧化物的无定形结构和缓慢的水解离阻止了氧渗透到Ti基底内部,抑制了Ti钝化,使Ir1O4阳极具有强大的稳定性。这些发现强调了表面氧化物结晶度对钛基阳极活性和稳定性的影响,并对如何调节单原子配位环境提供了见解,以获得更好的催化性能。
审核编辑:刘清
-
DSA
+关注
关注
0文章
49浏览量
15163 -
XPS
+关注
关注
0文章
97浏览量
12001 -
拉曼光谱
+关注
关注
0文章
83浏览量
2745
原文标题:上海交大JACS:单原子催化,非晶态载体更具优势!
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
奥拓电子吴涵渠董事长受邀出席上海交大活动
上海交大团队发表MEMS视触觉融合多模态人机交互新进展

使用Phase Lab2024A计算间隙原子扩散的非平衡凝固
宁德时代与上海交大共研多款机器人
SCR载体尺寸对背压及NOx转化效率的影响

桥田动态 桥田智能与上海交大达成产学研合作项目
上海交大国家卓越工程师学院莅临赛微微电子参访交流
非晶逆变器的弊端与优势有哪些
上海交大电院与奥拓电子共建智能视讯联合实验室
上海交大与云天励飞签署战略合作协议,寻找AI时代的Killer App

上海交大宣布突破:量子点液态生物芯片实现国产






 工商网监
工商网监
评论