 揭示固态电解质中锂盐解离的重要机制!
揭示固态电解质中锂盐解离的重要机制!
1、导读

清华大学深圳国际研究生院康飞宇(本刊主编)、贺艳兵(本刊执行主编)、柳明(本刊青年编委)、侯廷政(本刊青年编委)团队阐明了 BaTiO3铁电填料诱导 LiFSI解离的机制。通过引入不同极化态的 BaTiO3(BTO)填料,揭示了自发极化影响下 LiFSI 的解离机制。具有氧空位缺陷的四方 BTO3−x显著提升了 LiFSI 在 PVDF 复合固态电解质中的解离度,生成了 72% 的高浓度自由Li+,从而提高了离子电导率,达到 8.4×10−4S cm−1。该文章以 “Dissociation mechanism of lithium salt by BaTiO3 with spontaneous polarization”为题发表在Energy & Environmental Science上。第一作者是郭少柯和谭神冬。
2、研究背景
固态电解质搭配锂金属负极和高镍正极,能够实现高安全性和可达400 Wh/kg的能量密度。这使得它们成为电动车(EVs)和能量储存系统(ESSs)的理想选择。在这个过程中,固态聚合物电解质(SPE)由于其柔韧性和易加工性的特点,成为比固态无机电解质更有前景的选择。然而,传统聚合物基电解质在室温下的高结晶性阻碍了锂离子(Li+)的迁移,导致室温下难以运行。值得注意的是,在聚偏氟乙烯(PVDF)基聚合物电解质中,由于残留的N,N-二甲基甲酰胺(DMF)参与到锂离子的溶剂化结构中,削弱了锂离子与PVDF链之间的结合作用,因此,形成的[Li(DMF)x]+显著增强了PVDF基电解质在室温下的离子传输性能。但是,在PVDF电解质中,大多数锂离子以接触离子对(CIP)和聚集团簇(AGGs)的形式存在,而不是自由Li+。这种独特的溶剂结构为进一步提高锂离子传输效率造成了障碍。最近的研究发现,具有铁电特性的功能填料能够促使锂盐(LiFSI)解离,从而显著提高离子电导率。然而,要完全理解其内在机理却很困难,这阻碍了电解质性能的进一步提高。
3、工作要点
在这项研究中,作者选择了立方相钛酸钡(C-BTO)和四方相钛酸(T-BTO)作为代表性模型来探讨LiFSI在自发极化影响下的解离机制(图1 a,1b)。研究表明, T-BTO的偶极矩方向可以在电池的外部电场作用下略微偏转,以增强极化效果。沿着自发极化方向的T-BTO {001} 面展现出更加显著的LiFSI解离能力,并且能够吸附FSI−阴离子,这一效果通过引入氧空位(OV)缺陷得到进一步放大(图1c)。
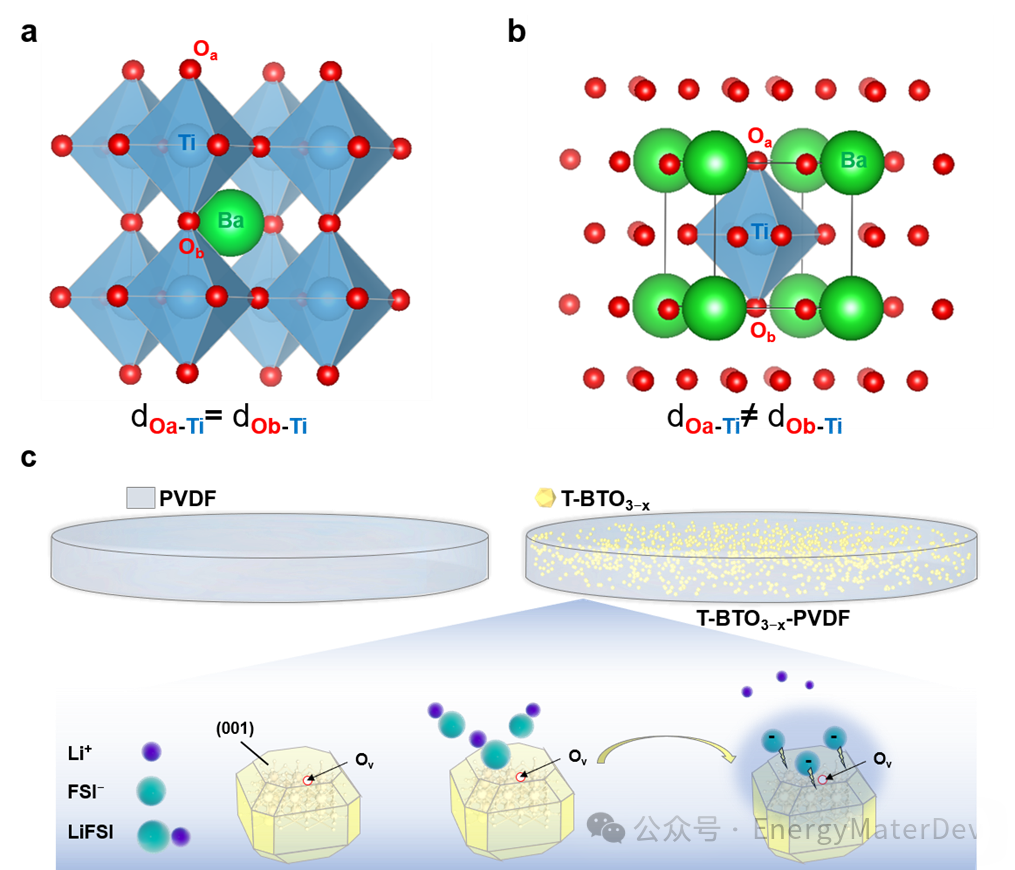
图 1 填料的晶体结构以及填料与 LiFSI 之间的相互作用过程示意图。a) C-BTO 的晶体结构,b) T-BTO 的晶体结构;c) T-BTO3−x的{001}面上 LiFSI 的解离和吸附过程示意图。
通过透射电子显微镜(TEM)图像,可观察到0.288nm和0.396nm的晶面间距,分别与C-BTO的(110)面和T-BTO的(100)面相吻合(图2a, 2b)。压电显微镜(PFM)展示了T-BTO的类矩形磁滞回线,表明其具有自发极化性质,而C-BTO的磁滞回线不明显(见图2c, S3)。为模拟电池环境中的实际极化情况,作者用薄膜铁电分析仪施加了与电池相当的外部电场(4V,80μm)。如图2d所示,T-BTO的磁滞回线未重叠并闭合,表明T-BTO中偶极子方向略微偏转,有助于极化的增强。为进一步增强自发极化,通过控制在H2-Ar混合气氛中烧结时间引入了氧空位,从而形成具有不同氧空位浓度的T-BTO3−x。T-BTO3−x的电子自旋共振(ESR)表明随着烧结时间延长, T-BTO3−x中氧空位浓度相应增加(图2e)。在没有LiFSI的T-BTO-PVDF薄膜中,其介电常数(εr)在10Hz时为46,在烧结6小时后,随着氧空位浓度的增加,εr最大可达59(图2f)。
此外,电子能量损失谱(EELS)提供了O-K(O1s 2p)微观结构信息,估计T-BTO3−x的表面氧浓度可以表示为T-BTO2.87(图2g)。研究中利用XRD的Rietveld精修揭示了T-BTO2.87的结构变化。在T-BTO和T-BTO2.87中的TiO6八面体中,dOa-Ti的距离从2.155Å增加到2.197Å,而dOb-Ti的距离从1.883Å减少到1.838Å(见图2h,2i,S5a,S5b)。dOa-Ti和dOb-Ti之间的距离差从0.272Å扩大到0.359Å。这表明由于氧空位的存在,Ti4+从TiO6八面体中心的电荷偏移程度显著增强,相应增加了自发极化。
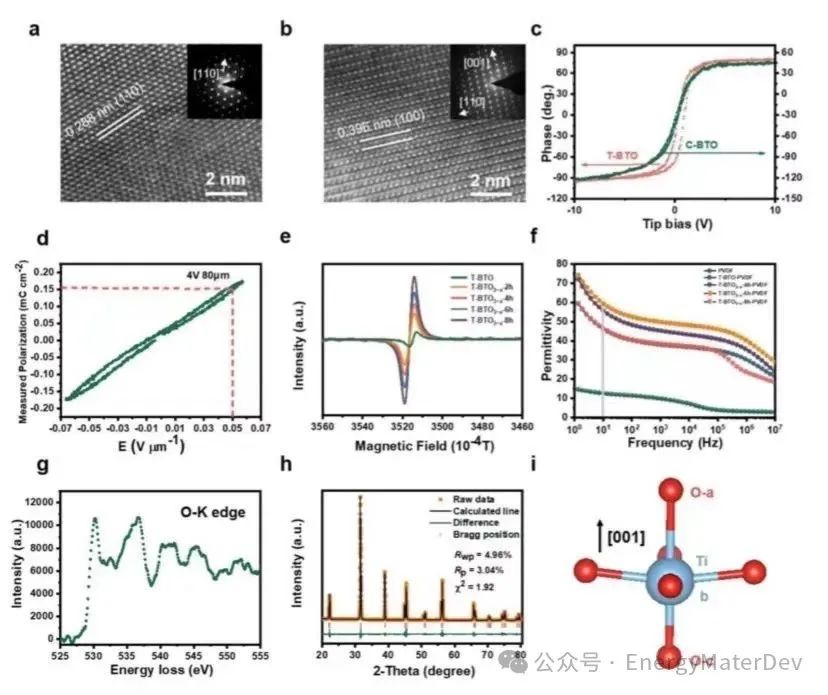
图2 C-BTO、T-BTO 和 T-BTO3−x的特性。a) C-BTO 颗粒和 b) T-BTO 颗粒的 TEM;c) 通过 PFM 分析获得的 C-BTO-PVDF 和 T-BTO-PVDF 的电滞回线;d) T-BTO 在电池电场下的铁电回线;e) 电子自旋共振,用于半定量氧缺陷浓度;f) PVDF、C-BTO-PVDF、C-BTO3−x-PVDF、T-BTO-PVDF和T-BTO3−x-PVDF的介电常数;g) T-BTO3−x O-K的电子能量损失图谱;h) T-BTO3−x的XRD Rietveld精修结果;i) T-BTO3−x的Ti4+偏移图。
为了进一步说明T-BTO与FSI−之间的相互作用,作者使用zeta电位在不同LiFSI浓度的DMF溶液中测试了填料的电荷状态。如图3f所示,在没有LiFSI的DMF中,C-BTO和T-BTO的zeta电位接近0。随着LiFSI浓度的增加,T-BTO的zeta电位显著下降,而C-BTO的zeta电位只轻微下降。由于FSI−是系统中唯一的负电荷粒子,这种差异表明,具有不同暴露晶面的T-BTO比C-BTO更有效地吸附FSI−(图3g)。通过绘制Wulff图,获得了受表面能影响的不同暴露晶面的分布(图3h)。因为C-BTO的晶体结构可以通过沿<001>方向拉伸扭曲为T-BTO的结构,因此它们的主要性质差异发生在{001}晶面上。通过使用DFT模拟C-BTO和T-BTO {001}晶面与FSI−的相互作用(图3h),可以观察到T-BTO {001}晶面显示出比T-BTO {100}和C-BTO {001}晶面更强的吸附能力(−0.360eV)。
因此,T-BTO {001}晶面在吸附FSI−中起着关键作用。为了更深入理解相互作用机制,作者进一步通过DFT模拟了锂盐的离解过程。如图3i所示,在体相中,Li+倾向于与FSI−的两个O形成双齿结构。然而,当LiFSI靠近T-BTO的{001}晶面时,FSI−的O倾向于与T-BTO的Ti结合,形成表面吸附的FSI−,从而使Li+从FSI−中解离。在表面引入氧空位进一步增强了这一现象。T-BTO3−x的zeta电位(−4.03mV)比T-BTO(−3.72mV)更负,且Li+的迁移数从PVDF的0.18和T-BTO-PVDF的0.29增加到T-BTO-PVDF3−x的0.52。
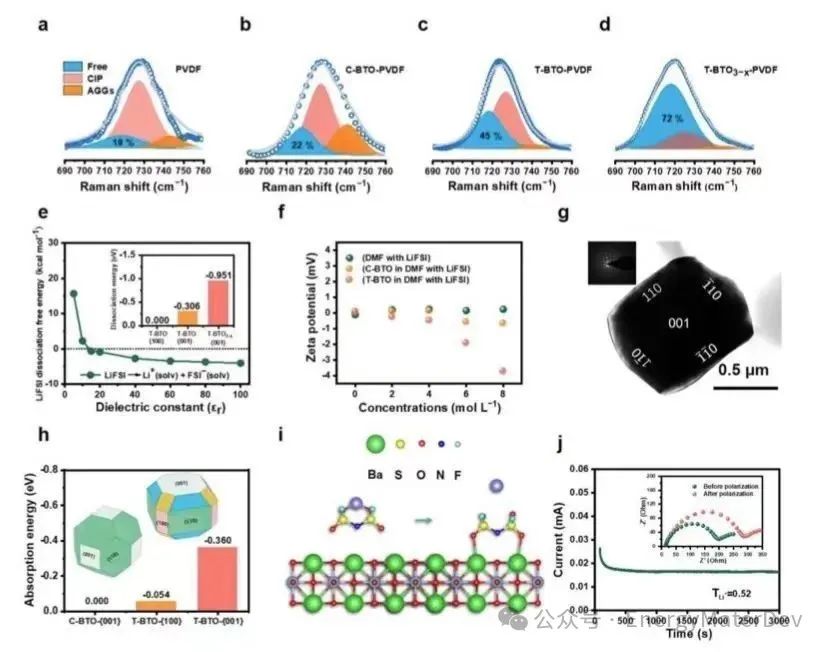
图3 LiFSI 的解离机理。a–d)不同填料电解质的拉曼位移;e)LiFSI 解离自由能与介电常数关系的理论计算;f)C-BTO和T-BTO 在不同锂盐浓度的 DMF 中的 Zeta电位;g)Z≈[001]方向的明场TEM和相应的选区电子衍射;h)不同表面对FSI-的吸附能计算;i) LiFSI 解离过程说明图;j) Li||T-BTO3−x-PVDF||Li 对称电池的电流–时间曲线和Li+迁移数。
纳米红外表明,PVDF 电解质中的DMF分布不均匀(图4a, 4b),而在T-BTO3−x-PVDF电解质中,DMF围绕填料均匀分布(图4c, 4d)。DMF的均匀分布有效地增强了Li+的传输,并减少了由于DMF的聚集而导致的界面副反应。此外,引入T-BTO3−x填料后,激活能(Ea)从0.3eV显著下降到0.179eV(图4e,S12)。因此,在25℃时,T-BTO3−x-PVDF的离子导电率达到8.4×10−4 S cm−1,而PVDF为4.2×10−4 S cm−1,C-BTO-PVDF为3.3×10−4 S cm−1,T-BTO-PVDF为6.5×10−4 S cm−1(图4f,S13)。
电化学测试揭示了T-BTO3−x-PVDF电解质在锂金属负极界面的优势。Tafel曲线显示,Li||T-BTO3−x-PVDF||Li电池的交换电流密度(j0=0.63 mA cm−2)明显高于Li||T-BTO-PVDF||Li电池(j0=0.31 mA cm−2)和Li||PVDF||Li电池(j0=0.05 mA cm−2)(图4g)。这表明在锂和电解质之间形成了更具动力学上有利的电荷转移界面。此外,填料可以将T-BTO3−x-PVDF的电化学窗口从3.7 V增加到4.4 V,这是由于其与高镍正极的优异兼容性(图S14)。随着T-BTO3−x的引入,T-BTO3−x-PVDF的拉伸强度从PVDF1.99MPa增加到2.45MPa(图S15)。
Li||T-BTO3−x-PVDF||Li对称电池的临界电流密度(CCD)可以达到2.2 mA cm−2,超过了匹配PVDF(0.6 mA cm−2)、C-BTO-PVDF(0.9 mA cm−2)和T-BTO-PVDF(1.4 mA cm−2)的电池(图4h)。此外,Li||T-BTO3−x-PVDF||Li对称电池在0.1 mA cm−2下显示了超过2000小时的长循环寿命)。值得注意的是,在0.5 mA cm−2的电流密度下,T-BTO3−x-PVDF的优势更加显著,表现出长达700小时的稳定循环时间,而其他电解质则不到210小时(图4i)。即使在电流密度为1 mA cm−2时,Li||T-BTO3−x-PVDF||Li对称电池也能保持稳定的循环超过376小时(图4j)。这些结果证实了T-BTO3−x可以极大地提高锂负极与T-BTO3−x-PVDF电解质之间的界面稳定性和动力学特性。
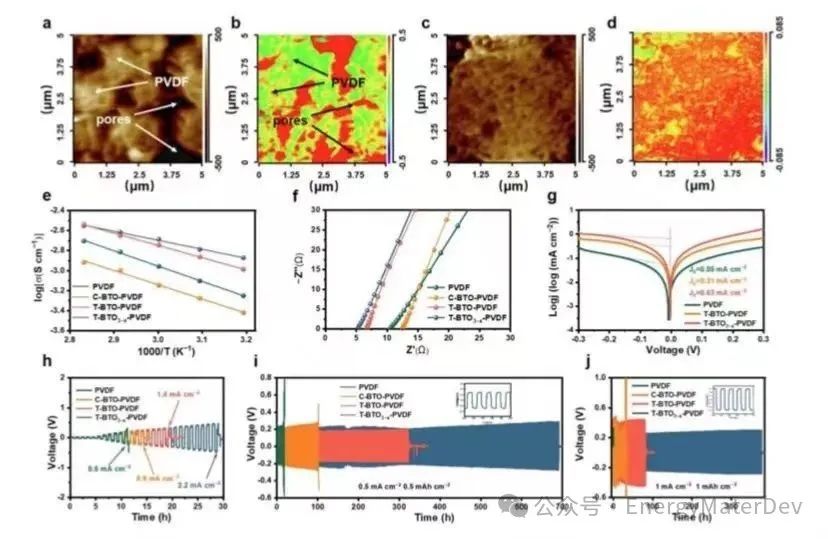
图4 T-BTO3−x-PVDF 电解质的特性。a) PVDF、c) T-BTO3−x-PVDF 的原子力显微镜图像,以及 b) PVDF 和 d) T-BTO3−x-PVDF相应的纳米红外图像(DMF 的 C=O 振动);e) PVDF 基电解质的Arrhenius图;f)钢钢(SS)||SS 电池的Nyquist阻抗谱;g) 不同电解质的Li||Li对称电池的塔菲尔图;h) 不同电解质的Li||Li对称电池的极限电流密度和不同电解质的Li||Li对称电池在i)0.5 mA cm−2-0.5 mAh cm−2 j)1 mA cm−2-1 mAh cm−2下的长循环。
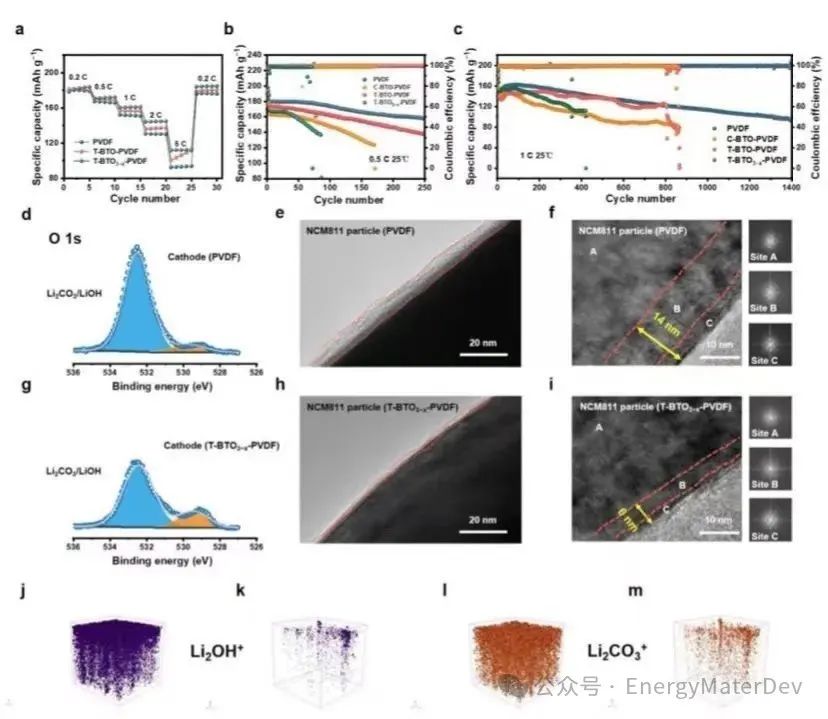
图5 NCM811||Li电池的电化学性能和界面表征;a) 25 ℃时,NCM811||Li电池的倍率性能;b) 0.5 C和c) 1 C 下的循环性能;d) PVDF和g) T-BTO3−x-PVDF的正极XPS图谱;循环后的NCM811颗粒与e, f) PVDF 和 h, i) T-BTO3−x-PVDF匹配的TEM图像。j, l) PVDF 电解质和k, m) T-BTO3−x-PVDF 电解质循环后锂金属表面Li2OH+和Li3CO3+的TOF-SIMS 3D重构。
电化学测试揭示了T-BTO3−x-PVDF电解质在锂金属负极界面的优势。Tafel曲线显示,Li||T-BTO3−x-PVDF||Li电池的交换电流密度(j0=0.63 mA cm−2)明显高于Li||T-BTO-PVDF||Li电池(j0=0.31 mA cm−2)和Li||PVDF||Li电池(j0=0.05 mA cm−2)(图4g)。这表明在锂和电解质之间形成了更具动力学上有利的电荷转移界面。此外,填料可以将T-BTO3−x-PVDF的电化学窗口从3.7 V增加到4.4 V,这是由于其与高镍正极的优异兼容性匹配(图S14)。此外,随着T-BTO3−x的引入,T-BTO3−x-PVDF的拉伸强度从PVDF(1.99 MPa)增加到2.45 MPa(图S15)。
进行了Li||T-BTO3−x-PVDF||Li对称电池测试以揭示BTO3−x的特殊影响。Li||T-BTO3−x-PVDF||Li对称电池的临界电流密度(CCD)可以达到2.2 mA cm−2,超过了使用PVDF(0.6 mA cm−2)、C-BTO-PVDF(0.9 mA cm−2)和T-BTO-PVDF(1.4 mA cm−2)的电池(图4h)。此外,Li||T-BTO3−x-PVDF||Li对称电池在0.1 mA cm−2下展示了超过2000小时的长循环寿命,而与PVDF、C-BTO-PVDF和T-BTO-PVDF相比分别为390小时、660小时和941小时(图S16)。值得注意的是,在0.5 mA cm−2的电流密度下,T-BTO3−x-PVDF的优势更加显著,表现出稳定的循环时间长达700小时,而其他电解质则不到210小时(图4i)。即使在临界电流密度为1 mA cm−2时,Li||T-BTO3−x-PVDF||Li对称电池也能保持稳定的循环超过376小时(图4j)。这些结果证实了T-BTO3−x可以极大地提高锂阳极与T-BTO3−x-PVDF电解质之间的界面稳定性和动力学特性。
速率性能方面,0.2 C 下,NCM811||PVDF||Li(179.7 mA h g−1)最初的放电比容量略低于NCM811||T-BTO3−x-PVDF||Li(185.8 mA h g−1)。随着速率的增加,NCM811||PVDF||Li的放电比容量(92.6 mA h g−1)远远落后于NCM811||T-BTO3−x-PVDF||Li(107.7 mA h g−1),表明BTO3−x(图5a)进一步改善了Li+的传输。当以0.5 C的速率循环时,NCM811||T-BTO-PVDF||Li和NCM811||T-BTO3−x-PVDF||Li在第250次循环时的容量保留率分别为88%和77%,而NCM811||PVDF||Li和NCM811||C-BTO-PVDF||Li电池分别在第86次和第170次时失效(图5b)。这表明T-BTO3−x通过调整溶剂化结构进一步改善了Li+的传输。此外,NCM811||T-BTO3−x-PVDF||Li电池可以在1 C下稳定循环1400次,容量保留率为61%(图5c)。与之形成鲜明对比的是,其他电池的循环次数不到900次,且放电容量更低。这些改进的性能归因于通过增加自由Li+而引起的离子导电率增加。
NCM811颗粒在1C下循环100次后的电解质界面(CEI)照片显示,与PVDF(7.2 nm)和T-BTO-PVDF(5.57 nm)相比,使用T-BTO3−x-PVDF的正极-电解质界面层更薄(2.6 nm)(图5e, 5h和图S19),岩盐相(C位点)和混合相(B位点)的厚度仅约为6 nm(图5i)。改善的溶剂化结构通过引入增强极化的填料减少了副反应,并增强了正极动力学。使用T-BTO3−x-PVDF的锂表面明显比使用PVDF和T-BTO-PVDF的锂表面更光滑(图S21)。循环后锂金属表面的飞行时间二次离子质谱(TOF-SIMS)的显示,与PVDF电解质(图5j, l)相比,使用T-BTO3−x-PVDF电解质的SEI中Li2OH+和Li3CO3+的数量明显减少(图5k, 5m),导致更稳定的界面。这些结果证明了T-BTO3−x的自发极化填料可以通过改善界面稳定性来增强电化学性能。
4、总结与展望
本工作重点是通过比较 C-BTO、T-BTO 和增强自发极化 T-BTO3−x来理解自发极化对 LiFSI 解离的影响。通过实验和理论模拟,研究人员发现 LiFSI 的解离程度与 T-BTO 的自发极化程度之间存在正相关。通过引入 Wulff 构造,研究人员提出了理论解释,证实了解离吸附机制中自发极化面的主导地位。这项研究为了解局部极化环境对溶剂化结构的影响奠定了坚实的理论基础,并为先进的复合固态电解质的设计提出了一种思路。
审核编辑:刘清
-
DFT
+关注
关注
2文章
232浏览量
22952 -
PFM
+关注
关注
1文章
157浏览量
28665 -
固态电解质
+关注
关注
0文章
86浏览量
5503
原文标题:清华大学康飞宇、贺艳兵、柳明、侯廷政最新EES:揭示固态电解质中锂盐解离的重要机制!
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
清华大学:自由空间对硫化物固态电解质表面及内部裂纹处锂沉积行为的影响
研究论文::乙烯碳酸酯助力聚合物电解质升级,提升高电压锂金属电池性能

陈军院士团队最新Angew,聚合物电解质新突破

一种薄型层状固态电解质的设计策略
半互穿网络电解质用于高电压锂金属电池
通过电荷分离型共价有机框架实现对锂金属电池固态电解质界面的精准调控
全固态锂金属电池的锂阳极夹层设计
固态电池中复合锂阳极上固体电解质界面的调控

无极电容器有电解质吗,无极电容器电解质怎么测
固态锂金属电池的外部压力研究

请问聚合物电解质是如何进行离子传导的呢?





 工商网监
工商网监
评论