 如何在化学和材料科学领域开展有影响力的人工智能研究?(一)
如何在化学和材料科学领域开展有影响力的人工智能研究?(一)
写在开头
近期,我有幸参加了多场既包括学术研讨又涵盖业界实践的会议,这些会议都集中讨论了人工智能与科学的结合。而通过这些深入的交流让我深刻体会到,在科学技术领域中,尤其是‘AI for Science(AI4S)’和‘Science for AI(S4AI)’这两个方向,人工智能的重要性正日益凸显,并且发展势头迅猛。为了更深入地理解这一领域的最新动态和应用,我投入了大量的时间和精力进行了学习和研究。在这个过程中,我意外发现了一篇名为《How to do impactful research in artificialintelligencefor chemistry and materials science》[1]的综述文章,它不仅提供了一个关于人工智能在化学和材料科学领域应用的全面的视角,而且还涵盖了当前的研究进展、面临的挑战以及未来的发展方向等内容。
文章首先深入探讨了人工智能在化学和材料科学领域中的应用现状及未来发展趋势。然后列举了机器学习在化学领域的多种应用,包括从预测分子性质到设计合成路线,再到模拟和分析,覆盖了化学研究的诸多方面。此外,论文还分析了机器学习研究者如何看待和解决具体的化学问题,其中将这些问题转化为机器学习的经典问题,如回归、分类、生成模型和智能体等,并探讨了基准测试、跨学科合作以及深度学习中的“惨痛的教训”等关键主题。
更重要的是,论文指出了选择有影响力的机器学习研究问题的标准,以及进行有效研究所需的步骤——包括数据收集、问题框架制定、方法选择和结果评估。文章强调机器学习在化学领域发展的三个关键方向:广度、深度和规模,并呼吁化学家与机器学习研究者之间的紧密合作,以解决更复杂和具挑战性的科学问题。
接下来,我将逐步分享这篇综述文献的内容。由于编译后的内容篇幅较长,因此我计划将其分为三个部分来逐一呈现。首先是第一部分编译的内容,如下:
第一部分编译后的内容:
摘要:机器学习已经广泛地影响了多个科学领域,包括化学和材料科学。尽管机器学习已经产生了显著的影响,但其潜力和成熟度尚未完全发挥出来。在这篇文章中,我们首先概述了机器学习在化学领域中各种问题在当前的应用情况。然后,我们讨论了机器学习研究者应该如何看待和处理该领域的问题。最后,我们提出了在化学研究中最大化机器学习影响的一些思考和看法。
1.引言
机器学习已经在化学领域得到了广泛的应用,并且这种应用正迅速增长。尽管如此,我们认为ML仍有更大的发展空间和应用潜力。目前的工作尚未充分发挥机器学习在推动化学理论和应用方面的广度、深度和规模潜力。此外,ML能够解决的实际问题类型,如假设生成或促进科学理解的内化,依然是活跃的研究领域或未解决的问题。
为了全面地了解这一领域,我们首先需要对化学问题进行分类,这些问题涵盖了预测、生成、合成、力场、光谱学、反应优化和基础模型等多个领域。接着,我们介绍了机器学习中涉及的问题类型,并展示了化学问题如何被重新构思为机器学习问题的实例。通过这些标准问题,有助于整理出机器学习提供的算法和理论工具。在深入探讨这一视角的基础上,我们还研究了机器学习和化学社区在实践和价值观上的差异,强调了合作和交叉启发的观点如何推动这两个领域的发展。有了这些基础,我们可以讨论如何选择在化学中应用机器学习的有影响力的应用,并推荐了我们在这一领域研究的良好实践。
2.数据驱动的化学:问题分类
对于化学,以及科学总体而言,都涉及以某种形式存在的数据。因此,可以毫不奇怪地认为,数据科学对化学是至关重要的。作为数据科学的一个子领域,机器学习已经成为我们学科领域不可或缺的工具。因此,对迄今为止的重要工作进行分类和组织变得尤其重要了。
我们提出了一种关于机器学习在化学问题中的应用分类法。如图1所示,机器学习被用于通过对化学结构、性质、3D结构与动力学以及实验数据进行编码和解码,从而解决各种化学问题。由于篇幅、时间和重点的限制,这并不是一篇全面的综述,而是一个强调机器学习在化学领域多样化应用的机会。我们不会详细介绍机器学习算法。如需详尽的评估,请参见其他文献。
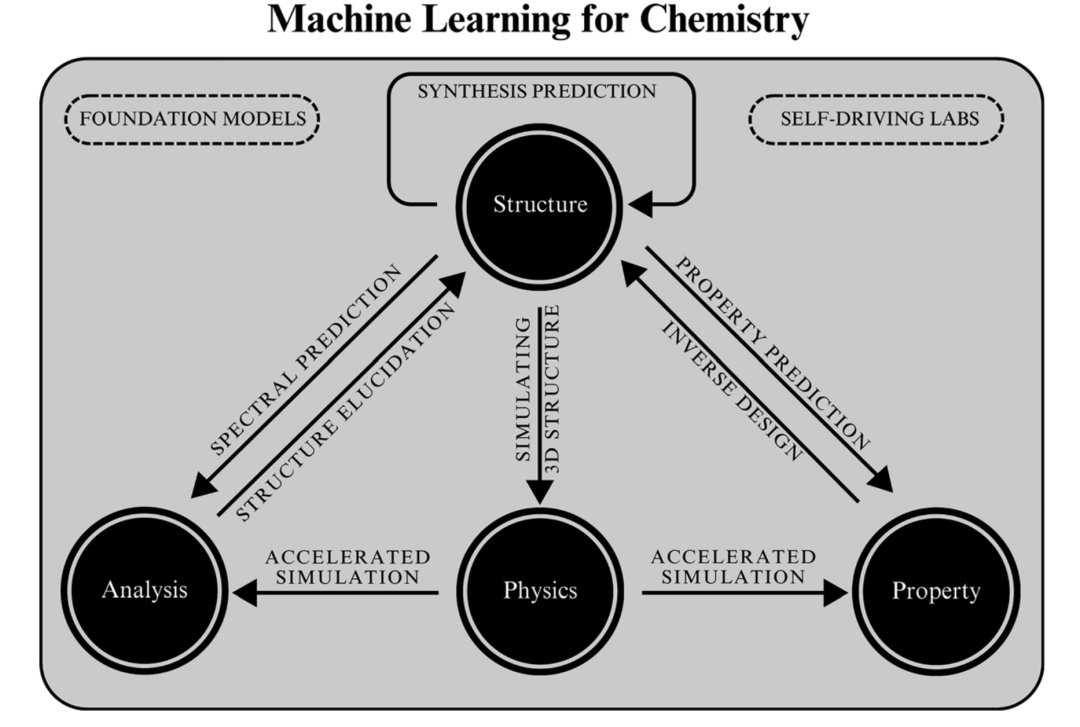
图1:与机器学习相关的化学问题分类法。每个箭头表示机器学习的应用,并展示这些内容之间的关系。基础模型和自动化实验室涵盖所有这些领域。
2.1 从结构到性质:性质预测
2.1.1 化学信息学与定量构效关系。
化学领域在“机器学习”这一术语广泛使用之前,就已经开始利用数据来预测化学结构的性质。这个领域最初被称为化学信息学。这些工具旨在存储、检索和建模化学结构。早期的例子可以追溯到1957年,当时进行的是数据库中的子结构搜索,随后便是简单的多元回归,用于学习定量结构-活性关系(QSAR),即分子描述符(如哈米特(Hammett)常数和分配系数)与生物活性之间的关系。这些研究主要集中在性质与活性之间的关系上——最早的结构-活性关系涉及局部解释,分析了环上的取代基如何影响活性,这种分析可以通过子结构分析推广到许多不同的骨架。最终,计算机自动将分子结构编码为指纹——一种位向量,用于存储分子中存在或缺失的多种子结构。这些指纹在编码分子结构以预测简单模型(如支持向量机)中的分子活性方面非常有用。
2.1.2 使用专家描述符表示分子
尽管化学家对官能团对分子性质的影响有一定的概念理解,但将这些信息有效传达给模型是确保模型具备预测能力的关键。专家描述符将来自实验或理论知识的化学信息注入到模型特征中,并在数据量较少的情况下取得了良好的预测性能。这些专家描述符在模型训练集之外也具有很好的泛化能力,因为这些特征蕴含了丰富的化学知识。早在1937年,哈米特(Hammett)就拟合了σ参数,以预测化学取代基对反应性的影响。此外,组分贡献方法假设结构组件或功能化在许多不同分子中表现相同,将这些组件参数化为可用于预测分子性质的数值特征。自那时起,该领域发展出了涉及分子指纹识别技术以及用于预测的二维和三维的信息。近年来,由于均相过渡金属催化剂的性质受到其附加配体强烈影响,因此对这些膦配体的结构特征和电子特征进行参数化也在催化剂性质预测方面取得了成功。回顾历史模型,最近的研究还成功地利用密度泛函理论(DFT)和机器学习来学习哈米特(Hammett)参数。
2.1.3 学习化学表示
随着计算硬件的进步,模型变得愈加复杂,从简单的线性回归模型发展到诸如自编码器、生成对抗网络、图神经网络和变换器等复杂架构。我们不再依赖化学家直观判断分子的最佳表示方式,而是可以利用模型自动学习并挖掘大量数据中的复杂模式,以进行性质预测。在一定程度的抽象下,这种方法往往忽视三维信息或波函数特性,分子可以自然地表示为图,其中原子是节点,键是边。通过将指纹的概念从离散比特向量扩展到连续特征向量,我们提出了图神经网络,以自动学习重要子结构的连续表征,并在分子性质预测任务中取得了最先进的性能。这些表征已广泛应用于多个领域,例如用于分子的嗅觉性质的机器学习,以及在催化中预测吸附物的吸附性质。
虽然构建图所需的简单原子特征和键特征可以迅速生成,但要进行预测的性质却更难以获取,尤其是在高质量和高准确度方面。由于学习到的表示通常需要大量的数据,因此复杂的架构在从典型实验设置中收集到的少量数据上表现并不理想。为了填补这一空白,研究人员创建了分子基准,以便能够正确评估这些学习到的表示的质量。这些基准包含了来自文献的数据任务,涉及预测生物行为以及物理化学性质或量子化学性质,并提供了一个共同的平台,使不同的机器学习架构能够以多种方式利用相同的数据进行性质预测。
为了提升图嵌入的性能,如果我们对如何调整嵌入空间以更好地反映输入之间的距离有一些直观理解,就可以进一步优化这些嵌入。这可以包括一些策略,例如使嵌入意识到化学反应如何转化这些嵌入,或者采用对比学习等方法。最后,对于那些对分子在三维空间中构象敏感的任务,将三维表示与固有不足的二维图结合起来,已被证明在预测分子性质方面是有效的。
2.1.4 限制和未解决的问题
尽管分子机器学习取得了巨大进展,但是机器学习模型在超出其训练数据范围时的泛化能力仍然有限,这给在新型化学领域应用带来了困难。有几种方法可能有助于弥补这些差距。例如,可以使用物理信息模型,这些模型能够包含一些基本表示,从而帮助模型更好地概括自身的表示,以满足与自然物理法则相关的一些对称性或特性。主动学习也是一种强大的工具,它可以通过捕获计算或实验数据来动态扩展数据集,以实现外推。此外,虽然模型在性质预测基准任务中的表现逐渐改善,但这些基准仅代表了化学任务的一个小子集,因此它们在其他具体任务上的表现仍不明确。尽管我们尝试创建更具代表性的基准,但这仍然不是社区的主要关注点。
结构-性质模型已被广泛应用于筛选项目中,并实现了一些经过实验验证的预测。我们将在第2.2.1节中讨论几个精选案例研
2.2 从性质到结构:设计分子的化学空间
理性设计范式分析了结构与性质之间的关系,以便设计出有前景的分子。而另一种范式提出的问题是:有哪些分子能够满足给定的性质?解决这个问题被称为逆向设计问题。
化学空间是所有可合成分子的集合,通常认为其规模庞大,至少有10^33到10^60个分子。在这片广阔的空间中,潜在药物可能治愈当前疾病,而某些假设材料则可能为实现可持续的未来提供支持。
2.2.1 虚拟筛选
在导航化学空间的过程中,一种简单的方法是首先列出一系列可行的选择,然后逐步缩减至最佳解决方案。为实现这种转变,实验上采用了高通量筛选和合成化学库中化合物的组合化学等策略。考虑到化学空间的巨大规模,随意搜索化合物只会产生少量有希望的结果,导致效率低下,因为广泛的化学合成活动的成本往往是高昂的或受限的。这促使了虚拟筛选和计算搜索漏斗作为一种方法,以过滤出不太有可能的化合物,仅留下最佳的候选化合物进行合成和测试。在药物发现中,计算机辅助的简单筛选可以排除高分子量或带有问题官能团的化合物,然后再进行更为计算密集的对接,以估计结合亲和力,最终缩小到少数先导化合物。随着虚拟库规模的扩大增加了有希望结果出现的可能性,这推动了越来越大规模的筛选活动,同时也需要越来越多的计算资源。例如,在哈佛清洁能源项目中,我们通过分布式志愿者计算进行了量子化学计算,搜索了10^7^个候选分子以寻找高效的有机光伏材料。
类似地,VirtualFlow通过高效利用数千个CPU核心对超过10亿个分子进行了对接。随着化学库规模的增长,所需的计算资源呈线性增加,而评估个别合成构件适应性的分层方法为突破线性扩展提供了一种途径。
2.2.2 生成模型的逆向设计
随着化学库的规模超过10^15个分子,筛选这些分子在计算上变得极为困难,机器学习(ML, Machine Learning)提供了一种在不模拟所有分子的情况下考虑大搜索空间的方法。例如,在一个化学库中,许多分子应该具有相似的结构和性质,因此对每个分子进行模拟是不必要的。处理这一问题的一种正式方法是先模拟库中的一部分,然后在这个子集上训练性质预测模型,这些模型应能够推广到整个库。由于这些性质预测模型的计算成本低于模拟,因此可以对整个库进行评估,并用于优先选择候选分子进行模拟。我们利用这一方法设计了经过实验验证的有机发光二极管(OLEDs)。
然而,机器学习的另一种方法则提供了一种考虑所有(或大部分)化学空间的方式。给定一个以SMILES字符串表示的分子数据集,生成模型学习生成与该数据集相似的字符串。由于生成模型可以考虑任意字符串,它们有潜力生成化学空间中的任何分子。同时,它们也可以被条件化,以生成具有所需性质的分子——本质上是逆转性质预测过程。分子生成模型已应用于多种模型类别,我们率先使用变分自编码器(VAEs)来实现这一目的。其他例子包括自回归模型、生成对抗网络(GANs)和强化学习等众多采样策略。此外,生成模型还扩展到各种表示形式,如SMILES、SELFIES,以及分子图和片段等。尽管遗传算法和贝叶斯优化等分子优化方法有时也被称为生成模型,但它们并不直接学习分子的分布。关于不同生成模型类别和表示形式的最新综述可见于Gao等人的研究[2],尽管这一领域正在迅速地发展。
随着越来越多的生成模型被提出,基准测试如GuacaMol和MOSES开始根据有效性、新颖性、独特性以及目标导向优化来评估和比较不同的生成模型。优化已成为主要焦点,因此可以将分子设计视为在分子图空间内对其性质进行组合优化。在这种背景下,一个新的基准强调样本效率,即达到最佳分子所需进行性质评估的数量。此外,我们最近在Tartarus基准集中提出了更现实的基准任务,这些任务依赖于模拟,更加贴近计算资源和实验资源受限的实际场景。
然而,当生成模型超越化学库覆盖整个化学空间时,它们放宽了合成性的关键约束。这意味着这些生成模型可能会产生一些难以合成和评估的分子。为了克服这个问题,可合成的生成模型在产生新分子时会考虑化学合成路径,从而确保所产生的新型分子不仅理论上有效,而且在实践中也是可合成的。另外,一些方法结合虚拟库与生成技术,以确保提出的新型分子始终来自于已有库。这些方法对于高通量阵列和自动化实验室尤其重要,因为如果预测出的新型分子无法在现有平台上合成,将可能延缓闭环方法的发展。
关于这些进展及当前领域内最新动态,Du等人提供了出色综述,总结了该领域最新的发展与方法。[3]
近年来,生成模型已证明其价值。其中值得注意的是InSilico Medicine公司利用这些模型成功开发了几种正在进行临床试验的新药。在2019年,我们与InSilico及中国无锡药明康德公司的研究人员共同展示了利用生成模型在约45天内开发出领先药物候选者的能力。从那时起,许多研究人员继续展示其他使用生成模型进行药物发现的新案例。例如,Barzilay及其同事采用类似的方法开发了抗生素。
2.2.3 限制和未解决的问题
虽然这样的模型能够轻松生成候选者,但候选者的质量取决于能否开发出一个表现良好且可扩展的成本函数,以对生成模型进行条件设置。此外,这些模型是基于近似度量进行训练的,这意味着它们在实际应用中的表现仍需评估。因此,评估候选者的可合成性或提供生成候选者的具体步骤至关重要(见下一节)。
大多数生成模型的开发通常以简单的基准为目标,比如预测log P这样简单的性质。然而,使用合适的基准(如Tartarus)进行开发,或将其限制在可合成的分子集合范围内,比如那些可以通过自动化实验室合成的分子(参见第2.7节),仍然是一个挑战。
2.3 从结构到结构:合成规划和反应条件预测
合成规划——即寻找能够生成理想目标分子的合成途径——是化学家们在过去一个多世纪中面临的一个持续挑战,尤其是在药物发现、农业化学或分子材料化学等“分子世界”领域。这个问题在两个方面都非常复杂:首先,给定所有反应物、试剂和反应条件,预测特定未见反应的结果至今仍然是一个未解决的问题。其次,即使拥有这样的“反应预测”工具,寻找可行的多步骤反应序列,以便最终从廉价且可商业获得的前体合成目标分子,还需要在庞大的可能途径网络中进行搜索。此外,合成规划问题还面临来自实际需求的额外挑战:效率、成本、废物产生、可持续性、安全性和毒性等都是在工业环境中特别需要关注的重要问题。
2.3.1 合成规划
合成规划通常采用逆合成的方法进行,这一方法由诺贝尔奖获得者E.J. 科里首创。通过利用化学反应性的知识,将目标分子逐渐被分解为越来越简单的前体,最终得到在市场上可购买的起始材料。形式上,这相当于一个树搜索问题。早在1960年代,科里就意识到这种方法非常适合以计算方式进行处理。从那时起,已经开发了多种专家系统来指导这一树搜索过程。
在过去十年里,利用机器学习工具箱应对这一挑战取得了显著进展。在这种情况下,关键的“决策策略”通常被视为一个多任务回归问题:给定目标分子的结构,训练一个机器学习模型以预测从反应目录中适用的反应。然而,这种符号化的方法需要一个预定义的包含所有反应类型的目录,通常被称为反应“规则”或“模板”,这本身又带来了新的障碍。关于“反应规则”这一术语,并没有一个普遍接受的定义,也没有明确的程序来从数据中提取反应规则。另一方面,“无模板”的方法针对一步反应预测问题,将反应预测视为起始材料图中的图编辑,或者解决一个序列到序列的“产品到起始材料”的翻译任务。值得注意的是,这些模型(无论是有模板还是无模板)可以在正向方向上进行类似的训练,即从起始材料预测反应产物。
这些单步预测模型用于构建树搜索模型,以解决综合规划问题。在这种情况下,蒙特卡洛树搜索(Monte-Carlo tree search)通常是首选方法。继Segler等人的开创性工作和Coley等人的研究之后,就已经发布了多个开源的系统。
2.3.2 反应条件的预测和优化
在合成规划中,常常被忽视的一点是,仅仅了解一种可能适用的反应类型,并不能保证所设想的中间体或目标产品能够从提议的起始材料中成功合成。产品是否能够获得(理想情况下是高产率),在很大程度上取决于通常所称的反应条件:试剂、催化剂、添加剂和溶剂的选择,连续参数(如计量比、温度和反应时间)的具体数值,以及实验室中进行反应时的实际操作细节。在理想情况下,一个人工智能辅助工具可以接收一个新的“起始材料到产品”的转化,并输出所需的反应条件。然而,这一目标尚未实现,主要原因在于反应条件涉及广泛的组合参数空间,并且通常受到难以模拟的基本物理原理的影响。在实际操作中,反应条件往往通过基于文献先例的“最近邻推理”来选择,这一过程可以是自动化完成,也可能依赖于人类专家的经验。
机器学习方法在反应条件优化方面主要集中于将反应产率作为反应条件的函数进行回归建模。在这一背景下,数据驱动方法与物理有机化学中的回归技术相结合,试图基于机理考虑来建模反应结果。在高度受限的条件空间中,利用高通量实验系统生成的数据进行的纯数据驱动监督学习在产物产率方面显示出了良好的效果。例如,我们在优化与制药工艺化学相关的反应E/Z比率的研究中发现,仅通过约100次实验,我们便超越了人类优化这一过程时所达到的先进水平。同时,利用文献数据进行相同目的的研究通常存在严重缺陷,这往往需要对每个案例进行单独的反应优化(更详细的讨论见下文)。在过去十年中,黑箱优化算法,尤其是贝叶斯优化(Bayesian Optimization, BO),变得越来越重要。在贝叶斯优化中,通过对现有数据进行贝叶斯推断来构建用于预测反应产率的概率模型。这些模型在整个优化过程中以迭代方式指导决策。而基于机器学习的替代模型进行迭代闭环优化的方法将在第2.7节中进一步讨论。对于条件优化,这些迭代方法在日益复杂的合成反应场景中表现出显著的效果。同时,化学领域的特定挑战,例如识别对多种底物“普遍适用”的条件,而不仅仅是针对一两个模型底物的条件,也推动了该领域算法的发展。值得注意的是,我们在铃木反应方面的研究使得条件具有普遍适用性,并且其产率是该领域之前先进水平的两倍。
2.3.3 限制和未解决的问题
尽管在过去十年中,基于机器学习的合成规划领域取得了显著的算法进展,但其实际应用仍然局限于开发相对简单的目标分子和短的合成路线。事实上,目前专家系统(即手动编码反应类型和适用规则的系统)仍然是计算机辅助合成规划的最先进水平。特别是,Grzybowski 的 Chematica 系统(现已商业化为 Synthia)在复杂自然产品合成或供应链意识的合成规划中展现了令人瞩目的实验应用。原则上,基于机器学习的算法应该能够提供与这些专家系统相似或更优的合成路线,但目前存在的问题主要归因于可用合成数据质量和数量上的不足,以及从数据中提取结构化知识时所面临的算法限制。我们和其他研究者最近对此进行了广泛讨论。
在反应结果和反应条件预测的背景下,类似的数据限制问题也被讨论过。专利数据以及商业数据库在数据报告的准确性、一致性或结构化方面存在严重问题,这不仅是由于错误数据、不一致数据或非结构化数据的存在,还因为在报告的实验中,人类偏见的影响,特别是对显著条件的偏重和对低产率记录的忽视,这些因素阻碍了从文献数据中进行反应产率预测建模。社区驱动的开源数据库,例如开放反应数据库(Open Reaction Database),代表了朝着更少偏见和更全面的数据收集迈出的重要一步——但这样的倡议需要在合成有机化学实验室中采用更为数字化的思维方式,以便更好地生成、收集和报告数据。
数据不足的另一个后果是缺少具有代表性的基准问题集。这种情况尤其适用于多步骤合成规划领域,在该领域中,迫切需要基准来进行合成规划性能的更为定量的评估。同样,化学反应性优化算法也需要具有代表性的基准,以评估标准贝叶斯优化算法如何适应化学反应性的复杂性。最重要的是,这些基准必须反映专家化学家所识别的现实问题,以激励和推动算法机器学习的进展,从而应对计算机辅助有机合成中的挑战。
2.4 物理结构:模拟和三维结构
机器学习使得数据驱动的解决方案能够应用于实验问题和计算问题。在有机化学中,分子的2D分子图结构是重点,而通过薛定谔方程,分子又建立在3D物理现实之上,这为预测分子性质和相互作用提供了丰富的量子力学和统计力学理论。模拟方法如密度泛函理论(DFT)和分子动力学(MD)可以利用这些理论来计算并预测分子的性质和相互作用。然而,尽管计算能力不断提升,这些模拟仍然计算成本高,这限制了它们只能在小系统和短时间尺度下进行。而通过从众多模拟结果中学习,机器学习提供了一个独特的机会来加速分子模拟。
2.4.1 神经网络势
量子化学中的一个基本问题是:对于给定一个分子,其在三维空间中表示为一组核点,如何求解薛定谔方程,并预测总能量及每个原子所受的力。力的计算使我们能够利用牛顿方程进行向前推进的动态模拟。然而,对于分子系统而言,求解薛定谔方程既复杂又计算成本高昂,而模拟牛顿方程则需要在每个模拟帧上计算力。因此,科学家们通过将简单函数拟合到实验数据来近似这些力,从而产生了第一个参数化力场,例如伦纳德-琼斯势。半经验模型结合了更多实验拟合参数,以便更准确地预测能量和力。这些经验力场使得经典分子动力学模拟成为可能,从而可以研究简单的蛋白质。然而,要捕捉像化学反应性这样的行为,则需要考虑量子效应。随着计算能力的提升以及更快的模拟方法(如密度泛函理论(DFT))的出现,最终使得在每个时间步上使用从头分子动力学求解薛定谔方程成为可能,但这需要付出巨大的计算成本。
神经力场的引入带来了显著变化。通过对密度泛函理论(DFT)数据进行训练,神经网络能够直接从三维核坐标预测能量和分子力,这使得分子动力学可以以从头计算的精度进行模拟,同时大幅降低了计算成本。由于分子力必须对分子的旋转保持等变性——也就是说,如果分子被旋转,分子力也必须“随之旋转”——这促使了保持这种对称性的等变神经架构的发展。在机器学习领域,神经力场经过了竞争性的基准测试,不断比较不同的架构和方法。Duval等人提供了这些等变架构发展的详细时间线。[4]随着能量和力的数据集不断增长,例如开放催化基准,神经力场也开始追求普适性了。
2.4.2 预测波函数和电子密度
相较于使用力场来预测能量,另一种预测能量的方法是直接预测波函数或电子密度。这种方法的优势在于,波函数和电子密度不仅包含了能量信息,还涵盖了系统的其他物理可观测量。例如,可以训练神经网络直接根据核坐标来预测哈密顿矩阵。对哈密顿矩阵进行对角化可以得到分子轨道,而这些分子轨道则构成了波函数。此外,可以利用预测的波函数来初始化自洽场迭代,从而加快量子化学计算的收敛速度。最近的研究表明,神经网络可以被训练,使其输出满足自洽性方程,这样就不再需要哈密顿矩阵的标签了。
此外,神经网络还可以作为假设形式,直接表示波函数。在这种情况下,网络以电子坐标为输入,并输出波函数幅度。采用相同的随机优化算法,神经波函数可以被训练以最小化变分能量并满足薛定谔方程。这种方法最近也扩展到了激发态。
另一方面,在密度泛函理论中,也可以训练神经网络直接根据给定的核坐标来预测电荷密度。同时,机器学习也被应用于学习密度泛函。
2.4.3 预测和生成3D结构
即使存在快速准确的力场,许多问题仍然依赖于找到分子的能量优先构象。然而,构象空间依然非常庞大,特别是对于大型体系如蛋白质而言,实际上是无法穷尽的。同样,在建模化学反应时,庞大的构象搜索空间使得识别过渡态变得具有挑战。为解决这些问题,机器学习方法可以直接预测和生成3D结构。
大规模构象搜索空间的复杂性促使生成模型用于引导这一空间的探索。无条件生成模型,如等变扩散模型,能够同时生成3D原子位置和原子类型。在解决寻找给定分子稳定的3D构象问题时,原子类型可能保持不变,生成则是基于2D分子图的条件。一些方法可自由生成原子位置,而其他方法则生成可旋转键的扭转角度。最近的研究显示,放弃扭转和旋转对称约束可能带来更好的结果,尽管成本更高。有一个相关的任务被称为对接,即在蛋白质口袋内进行配体的构象搜索,以评估结合亲和力。这一过程也已经使用扩散模型来来研究了。
在晶体结构预测问题中,目标是找到给定组成的最稳定周期性原子排列方式。传统方法是通过搜索所有稳定的原子坐标和晶格矢量的排列组合来找到能量最低的结构。而等变扩散模型则为这一问题提供了自然的解决方案,它可以同时扩散坐标和晶格参数,并强加空间群约束以进一步提升性能。实际上,将这种扩散方法扩展到大型数据集,使逆向设计能够同时满足多个期望的性质。
在与生物分子模拟相关的领域中,三维结构预测问题普遍存在。长期以来,从蛋白质序列预测折叠的三维蛋白质结构这一难题在一定程度上已通过AlphaFold及相关模型得到了有效解决。在此基础上,扩散模型生成了以刚性残基序列表示的蛋白质骨架。这些模型取得了显著成功,甚至被用于设计满足结构约束的蛋白质,并经过了实验验证。这些扩散模型的应用范围已扩展到所有生物分子,其方法能够预测蛋白质、RNA、DNA和配体如何在三维原子细节中的组装方式,从而涵盖了对接任务,因此有望在未来成为药物发现的重要工具。
2.4.4 增强采样和粗粒化模拟
寻找最稳定的几何形状固然有其价值,但要真正模拟分子之间的热力学相互作用,则需要对三维结构的平衡分布进行采样。平衡态遵循与能量相关的Boltzmann分布,而学习这种平衡分布的生成模型被称为Boltzmann生成器。深度生成模型开始通过流匹配这一扩散模型的变体来解决这个问题,并且已经在多种不同类型的肽中证明了其可转移性。另一种方法则是利用福克–普朗克方程来学习如何采样平衡分布。
在粗粒化过程中,通常将原子聚集成所谓的珠子,这样就可以降低计算成本,并且能够捕捉到长时间尺度的事件。然而,这些粗粒化珠子的力需要与全原子力进行拟合。为了解决这个问题,可以应用神经网络来学习粗粒化力场,通过预测自由能的梯度,而不是能量,并将这些预测的力与全原子力进行匹配。而使用流匹配方法则消除了对全原子力的需求,仅需粗粒化珠子的平衡样本即可。此外,扩散模型可以同时学习生成模型和粗粒化力场。
虽然粗粒化力场的评估速度明显快于原子级力场,但分子动力学模拟仍然受到必须使用飞秒级积分时间步长的限制。为了解决这一问题,平衡方法的替代方案主要集中在加速分子动力学,以实现更长的时间尺度。
这可以通过“时间粗粒化”来实现,即训练生成模型以预测在较大时间步长下的结果。此外,研究还致力于将模型扩展到多个热力学性质范围,如温度和压力。这使得能够模拟不同环境,并对之前不适合的数据进行训练。通过在模型输入中添加温度等额外参数,可以将相应的粗粒化自由能函数的导数纳入损失函数中。而自由能的高阶导数作为响应性质,可以通过多次反向传播计算得到。因此整合热力学参数可能是整体模拟生物或工业环境的重要因素之一。
对于稀有事件采样,例如化学反应和过渡态搜索,已经出现了无反应坐标的过渡路径采样方法。另一方面,当反应物、产物和过渡态的数据集可用时,可以直接训练生成模型,进而可以基于反应物和产物生成过渡态。
2.4.5 限制和未解决的问题
虽然神经力场可以达到很高的准确性,但它们仍然需要足够的训练数据来覆盖整个相空间。如果没有完全覆盖,神经力场可能会陷入不稳定的动力学状态。一项基准测试强调,评估力场的标准应基于其动力学表现,而非力的误差。
然而,随着神经力在越来越大数据集上的训练,这些问题可能会逐渐得到解决,从而推动通用力场的发展。尽管机器学习模型受限于数据质量,但新数据可以通过模拟生成,这为数据可用性和大型模型的发展带来了乐观前景。
与此同时,要在大长度和时间尺度上进行有效模拟仍需大量工作。在亚稳态条件下进行适当平衡采样面临着重大挑战,相关的稀有事件采样问题也依然是亟待改进的领域,因此成为了近期众多研究工作的重点。
2.5 结构和分析:光谱学和结构解析
在化学领域,一个自然而又未被充分利用的机遇是利用机器学习来进行结构解析,它旨在通过光谱或其他分析数据预测二维或三维分子结构。就像计算机视觉使得计算机能够感知自然界一样,计算光谱学也可以让机器通过分析仪器感知分子世界。随着实验自动化技术的进步,预计将合成越来越多的从头合成和未知的化合物,这推动了对更快且准确的结构解析需求,以充分支持这些自主分子发现和反应发现平台。
2.5.1 正向光谱预测
数据驱动的结构阐明最直接的方式是存储一个光谱库,针对给定的光谱在库中搜索匹配项,然后检索相应的结构。为了扩大光谱库的覆盖范围,可以使用前向光谱预测来为特定化学结构预测其光谱。尽管物理模拟提供了一种有根据的方法来预测光谱,但其过程往往复杂且计算成本高昂。另一种方法则是利用机器学习从结构出发预测各种类型的光谱,包括质谱(MS)、核磁共振(NMR)和紫外-可见光谱(UV-vis)。一些研究将前向预测问题视为公式预测,采用自回归模型或固定的公式词汇;而另一些研究则专注于子图预测,利用递归分解、自回归生成和深度概率模型,或结合三维结构信息。在质谱的背景下,一些方法将光谱近似为具有相应峰强度的离散区间,从而简化问题,将其转化为直接从结构回归质谱的任务。除了结构到光谱的预测外,另一种方法涉及通过估计各种分子描述符来预测结构-性质关系——这些描述符可以是标量(例如能量、部分电荷)、向量(例如电偶极子、原子力)以及高阶张量(例如Hessian矩阵、极化率、八极矩)——然后利用这些描述符来预测不同类型的光谱,包括红外光谱、拉曼光谱、紫外-可见光谱和核磁共振。
2.5.2 结构解析
直接从给定光谱预测化学结构的过程被称为逆问题。DENDRAL是第一个用于从质谱推断化学结构的专家系统,诞生于1969年。化学家们还利用机器学习(ML)分析红外光谱(IR)、核磁共振(NMR)和质谱,以识别有限的官能团。虽然这些方法提供了有用的结构见解,但它们无法完全解析分子结构。
结合多个推断出的官能团的信息,使得结构解析成为可能。对于NMR数据,分子结构可以通过首先识别分子亚结构和官能团来进行解析,然后通过波束搜索在可能的配置中进行最优组合,或者逐个原子构建,这与化学家在解释NMR光谱时采取的方法相似。类似的“亚结构重建”策略也在红外光谱和表面增强拉曼光谱(SERS)中以不同程度的细节应用。然而,随着原子数量的增加,这种方法很快会遇到组合规模问题。
分子结构解析可以从深度学习的角度将其视作为一个端到端的问题。在这种方法中,光谱被标记为字符串,并预测SMILES字符串;这可以视为一种机器翻译任务。这种方法已经应用于核磁共振(NMR)、红外光谱(IR)和串联质谱(MS/MS)数据中了,显示出来在扩展到更大化学系统和从头结构解析方面具有更显著的潜力。此外,结构预测问题也可以被表述为一个优化任务,例如,可以将其形式化为马尔可夫决策过程。如果我们考虑一些关于当前化学系统的先验信息,如化学式、已知的起始材料和反应条件,将这些信息作为约束条件应用,就可以帮助模型更有效地收敛到一个解决方案。
随着研究的深入,从分子到晶体的转变,解决粉末X射线衍射(PXRD)和X射线吸收近边结构(XANES)等X射线光谱数据的逆问题,为机器学习领域带来了新的挑战。这为不同深度学习模型在晶体系统和空间群识别中的应用提供了独特且尚未充分开发的机会。特别是,扩散模型展现出了良好的前景,尤其是在文本到图像生成这一对应逆问题中的成功应用。在这个背景下,我们可以在文本与光谱之间,以及图像生成与晶体结构预测之间建立类比。
在旋转光谱学领域,光谱分配的挑战——即从密集的旋转光谱中推导旋转常数——代表了机器学习在这一领域早期应用之一。由于光谱密集且易于模拟,这个问题特别适合深度学习技术。然而,仅依靠旋转常数无法确定分子的三维结构。我们最近提出的方法通过推断三维结构来解决这一问题,所需的信息包括分子式、旋转常数以及称为替代坐标的无符号原子笛卡尔坐标。
在结构生物学领域,蛋白质结构预测的进展与冷冻电子显微镜技术的发展相辅相成。利用冷冻电子显微镜重建蛋白质结构的过程已经采用了深度生成模型。这些方法已发展到能够从冷冻电子断层成像(cryo-ET)中重建生物分子动态的程度。利用冷冻电子显微镜进行的结构解析工作也在不断取得日新月异的进展。此外,数据处理方面的进步使得分辨率得到了显著提升,而这些改进都可以通过使用机器学习方法进一步增强。
2.5.3 限制和未解决的问题
与所有需要大量数据的方法一样,一个关键问题始终存在:虽然可以获得大量的模拟光谱,但考虑模型在实验光谱上的表现是否令人满意至关重要,因为实验光谱往往表现出更大的变异性和不一致性。一个值得思考的问题是:科学界是否应该更加努力推动将原始光谱文件存放在开放数据库中,以促进深度学习在从自动化光谱到结构解析中的应用?
对于逆向光谱到结构解析的工作中,虽然纯样品的自主分子结构确定无疑对高通量反应优化和发现活动至关重要,但同样重要的是解决来自复杂混合物的光谱结构的注释问题,这包括对特定目标化合物的定向识别和非定向代谢组学。这类混合物在实际样本矩阵中是常见配置,对于生物诊断、法医学等多个领域都是不可或缺的。这些任务的成功在很大程度上依赖于模型从复杂的数据中解开并分离出单个分子光谱特征的能力。机器学习擅长处理复杂、高维数据,因此非常适合处理这些具有挑战性的任务。此外,利用机器学习方法集成来自多个光谱输入的信息,可以进一步提高结构解析的准确性和完整性。
2.6 利用基础化学模型实现规模效益
随着计算能力的提升,机器学习模型已经在越来越大规模的数据集上进行了训练。在大规模应用中,机器学习展现出不同层次的能力。而基础模型是经过广泛数据训练的大规模模型,能够应用于多种下游任务。一些通用基础模型,如ChatGPT、Gemini和Llama,通常用于语言生成和图像生成;其中许多模型仅针对语言进行训练,或者是在多模态上进行训练。然而,在化学领域使用这些模型面临着独特的挑战,因此许多研究者选择从头开始在化学数据上训练他们的模型,但这并非易事。在本节中,我们将探讨化学领域基础模型的现状,并提出对尚未解决问题的看法。
2.6.1 通过大型语言模型和智能体进行知识转换
一些最早将生成模型应用于化学的研究是通过语言实现的,这得益于分子可以使用SMILES表示法以字符串形式进行表示。早期的化学语言模型是通过无监督学习对SMILES表示进行训练的,它学习了分子子片段之间的依赖关系。最近,这些模型也开始同时在其他以文本标记表示的分子形式上进行训练,例如文本描述、科学论文和合成程序,它们通常采用自回归损失,以便在推理时能够生成分子的描述或结构。Ramos等人撰写了一篇综合性综述[5],详细介绍了迄今为止80个化学/生物化学语言模型,以供读者进一步了解。将文本描述纳入模型的一个动机是,它们包含有关分子功能性质的信息,这对于改善结构相似但功能不同的分子的嵌入表示非常有帮助,反之亦然。这些文本描述还让用户能够使用自然语言与模型互动,这比起僵硬的查询方式对许多用户来说更加直观。此外,大型语言模型(LLMs)还被用于科学文献解析,促进从现有文献中提取化学信息并构建知识数据库。这些数据库可用于对LLMs进行微调,有潜力提升自动化实验室在生成和筛选方面的能力。
然而,使用这些模型进行开箱即用的发现任务或特定领域的化学应用仍存在一定差距(至少根据我们的了解)。造成这种情况的原因之一是,缺乏足够的数据来以与GPT-4等模型在网络规模文本和图像上训练相同的方式对这些模型进行训练。利用这些具备化学知识的语言模型的一种方法是对其进行微调,以适应下游任务,或者将其嵌入到优化框架或搜索框架中,从而提供良好的先验知识。其他研究也开始了探索模型和数据的扩展。
化学感知基础模型的一个有趣应用是开发化学智能体,这些智能体能够利用解决化学问题所需的工具,或规划化学实验。一些显著的例子包括ChemCrow、Coscientist、我们自己的ORGANA和ChemReasoner。这些智能体可以访问各种与化学相关的工具,如模拟器或机器人,以执行化学实验,并使用大语言模型(例如GPT-4)作为中央协调者,决定何时以及如何使用这些工具来实现用户指定的目标。此类智能体的一个长期目标是开发科学助手,帮助进行更复杂的推理和规划,通过自主生成和完善假设,超越单纯的计算和执行。AI科学家将这一理念扩展到了其他研究领域,展示了通过执行实验和撰写研究论文进行自主机器学习研究的能力。
这些研究领域仍处于起步阶段,因此存在几个未解的问题,包括:(1)我们如何有效地评估化学感知大语言模型(chemistry-aware LLMs)/智能体(agents)?(2)这些模型在化学家实际工作中的使用案例是什么?有效的模型评估主要依赖于开发有意义的任务,而这在数据集的规模和广度上目前仍然是一个开放性问题。虽然在这一领域已经存在几个基准测试,这为我们提供了一个良好的起点,但在数据质量和任务目标方面仍有改进空间。最近发布的一些基准测试更接近实际应用,同时,像Polaris这样的平台使研究人员能够更快速地访问各种数据集。然而,在这一领域使用次优基准测试的问题因当前机器学习环境而加剧,因为基准测试主要用于展示新方法相较于现有最优技术所取得的更好表现,但人们并不理解其改进的原因。这也为化学家与机器学习领域专家社区之间合作的提供了一个绝佳机会。
基于语言的基础模型也被应用于其他领域,包括知识图谱生成和从化学文献中进行知识提取,其中还包括我们在反应图解析方面的工作,这是一项具有挑战性的任务。这些努力对于创建结构化的实验程序数据库是至关重要的,这些数据库可以为现有的资源库做出贡献,例如之前提到的开放反应数据库。
2.6.2 基础物理模型
虽然仅语言基础模型在化学领域受到广泛关注,但研究表明,语言可能并不是唯一有效的手段,特别是在三维几何结构至关重要的情况下。例如,Alampara等人显示,仅依靠语言模型无法编码表示特定材料属性所需的结构信息。
然而,在生物化学科学中,语言模型并不是唯一开发的基础模型。在已经构建了几种模型中,这些模型能够对力场进行普遍近似,并预测任何分子、材料或蛋白质的结构。或许最著名的例子是用于蛋白质结构预测的AlphaFold2,以及最近推出的AlphaFold3,该模型能够根据任何一组二维生物分子预测它们在三维中的组装方式。据我们了解,这些模型在许多结构和功能任务中仍然优于任何基于序列的蛋白质预测模型,尤其是在输入序列在训练数据中没有同源物的情况下。
另一个引人注目的例子是最近推出的基础模型MACE-MP-0,该模型采用了MACE等变架构。其MACE-MP-0模型在150,000个无机晶体上进行了训练。在经过少量特定任务示例的微调后,它可以作为力场用于多种任务的模拟,甚至包括一些看似毫不相关的小蛋白质模拟。值得注意的是,MACE-MP-0中的分子间相互作用似乎存在一定的不明确性。例如,在前面提到的蛋白质模拟中,该模型能够成功捕捉氢转移,这是一项显著的成就。然而,作者也选择加入了源自经典计算化学的D3色散,这表明该模型在预测长程相互作用方面仍然需要一些帮助。值得一提的是,基础力场的研究也正在持续扩展,如工业研究实验室正在越来越大的数据集上训练神经力场,例如GNoME和MatterSim。
从这些模型中得出的一个关键结论是,结构信息不应被忽视,这取决于模型所应用的下游任务。同时,在广泛的大规模数据集上训练模型(即超越仅在单一预测任务上训练简单模型的传统,这在几年前仍然是常态)可以更好地推广到更多的下游设置中。我们认为,同时在多个模态上进行扩展对于构建化学领域中最佳的基础模型至关重要——即尽可能多地在各种模态上训练模型,例如三维结构信息、文本和光谱信息。
2.6.3 限制和未解决的问题
在领域科学的研究中,我们在数据条件上并不如自然语言或图像领域那样优越,因为后者已经拥有了网络级的数据可供利用。而科学数据则相对稀缺;每一个数据点必须通过实验获得,或者是通过高质量的模拟得出的。如果采用模拟方法,模型必须找到将其结果转化为特定实验条件的方法。我们怀疑,跨化学领域的通用模型仍然需要十年的时间才能实现,并且随着人类对这些模型需求的不断增加,这一目标可能会变得更加难以捉摸。这就像扩宽高速公路的问题一样,许多分析师已经表明,一旦道路被扩宽,由于其可用性而产生的额外需求会立即导致高速公路交通拥堵。
2.7 闭环优化和自动化实验室
2.7.1 自动化实验室
随着机器学习应用的不断发展,高质量数据的必要性和稀缺性变得愈加明显。化学数字化的兴起以及机器学习的进步,为将机器学习与通过机器人实验进行自动数据生成相结合奠定了基础。这种协同作用催生了自动化实验室(SDL)的概念。自驱动实验室主要由两个关键组成部分构成:自动化实验室设备和实验规划者,这两者都利用机器学习技术来提升其功能。最终目标是自主执行科学方法,包括假设生成(机器学习)、假设测试(实验)和假设细化(机器学习),从而以数据高效的方式探索广阔的设计空间。
通过将机器学习与计算机视觉相结合,自动化实验室设备取得了显著进展,形成了“通用化学机器人”的概念。这些经过机器学习训练的机器人能够基于外部反馈做出决策,从而实现传统上由人类化学家执行的化学操作的动态自动化。鉴于在基于外部反馈进行主动决策的机器人设备训练中固有的挑战,该领域的一个显著创新是利用数字孪生体——实验室设置的虚拟复制品——为加速机器人机器学习模型的训练提供了坚实的框架。这些数字孪生技术通过高精度模拟化学场景,建立了一个真实的反馈机制,加速了模型的学习过程。
在实验规划方面,启发式技术正在逐步被机器学习优化算法所取代。当这些优化技术与化学数字化相结合时,它们能够识别目标化学物质并优化反应条件,同时显著减少所需的实验步骤。在各种机器学习优化技术中,贝叶斯优化因其在化学应用中的成功而在实验化学领域尤为突出。基于机器学习的代理模型能够预测化学物质和反应的性质,这在过程优化和材料发现中发挥了重要作用,并且已有许多成功案例记录。
此外,大型语言模型(LLMs)的兴起进一步增强了自动化实验室(SDLs)的辅助组件。而大型语言模型被有效地用于创建无需传统编码的人机界面,使得化学家与实验室系统之间的交流更加自然。这对于那些不太擅长编码或数据处理的用户来说,是一个显著的优势。
2.7.2 限制和未解决的问题
正如我们最近讨论的那样,自动化实验室系统面临的挑战大致可分为两大类:自动化(硬件相关)和智能化(人工智能相关)。
自动化挑战。主要的硬件挑战源于化学仪器设计时更偏向人性化设计,并未考虑自动化需求,以及与现有自动化模块之间缺乏无缝连接。因此,大多数自动化实验室系统是以半自动方式运行的,需要人工干预进行诸如样品转移、维护和故障排除等任务。已经提出了各种解决方案来解决这些问题,包括部署移动机器人进行样品转移,以及调整通用机器人来执行化学任务或操作最初设计用于人类使用的仪器。然而,这些方法大都依赖于传统算法,需要静态校准,因此并不太适合自动化实验室系统的动态特性。虽然已经提出了将计算机视觉和人工智能相结合作为解决方案,但实验室设备,特别是玻璃器皿,仍然面临显著的挑战,不过这些挑战正在逐渐得到解决。
认知挑战。认知挑战主要在于开发能够准确估计系统输出的模型,这一困难限制了更通用生成模型的应用,有效地降低了实验规划者在化学领域中所能探索的空间。结合前面提到的挑战,一个明显的问题就是:SDLs通常在数据匮乏情况下运行。而预测和生成的机器学习模型通常需要大规模的数据集才能做出有意义的预测。虽然生成模型可以在现有数据上进行训练,但在这种低数据情境中部署预测算法仍然是一个重大挑战。
辅助组件的挑战。在关于自动化实验室系统的辅助组件方面,LLM的整合显示出在自动化工作流程创建和改善人机交互方面具有潜力。然而,需要进一步研究以确保这些过程的安全性和可靠性。此外,虽然将文献提取整合到自动化实验室中可以增强模型开发,但其与预测模型的有效整合仍然是一个未解决的问题。
在自动化实验室领域,需要解决的最后一个挑战是发展规模经济的问题。随着社区建立的自动化实验室数量的增多,建立下一个自动化实验室的难度将会大大降低。因此,实现低成本自动化实验室的民主化对该领域的发展来说至关重要的。
写在最后
在读完这篇文献后,我深刻意识到,个人在追求最大化影响力的过程中,有两个关键能力至关重要。首先是会发现问题,以及能判断出哪些问题值得解决的能力;其次是具备高质量解决特定领域问题的技术水准。
在学术研究中,发现问题的能力体现在研究者能够敏锐地识别出关键的科学问题,并判断其研究价值如是否能引领新领域的探索。而具备高质量解决问题的技术水准,则体现在研究者能够运用先进的实验设计、数据分析和建模技术,去有效应对复杂的科学挑战。结合这两种能力,研究者不仅能够提出创新的理论,还能够产生具有实用意义的科研成果,促进知识的快速积累与传播。
在技术开发方面,发现问题的能力体现在开发者能够识别出行业内最迫切需要解决的技术问题或瓶颈,判断其对产品和服务的影响。而具备高质量解决特定领域问题的技术水准,则表现在开发者能够运用工程原理、编码能力和系统设计知识,去创建和优化技术解决方案。有效的技术开发需要两者的结合,以确保所开发的技术不仅具有实用性,还能够在市场上产生积极的反响。
在产品开发方面,发现问题的能力体现在团队能够识别用户需求、市场趋势以及潜在的痛点,判断哪些问题最值得优先解决,以增强产品的竞争力。而具备高质量解决问题的技术水准,则体现在团队能够将创新的技术和设计理念转化为可行的产品特性,从而有效满足用户需求并实现商业价值。只有结合这两项能力,团队才能够更好地开发出符合市场需求和技术标准的产品,从而推动业务增长。
虽然具备高水平技术解决问题的人数远远多于那些能够识别真正有价值问题的人,但真正能够在各个领域产生深远影响的,通常是那些同时具备这两项能力的人。因此,每个从业者都应时刻思考“什么才是值得解决的问题”,而这一思考将成为我们在学术、技术或产品发展中追求创新与影响力的核心驱动力。
参考文献:[1]
Spiers Memorial Lecture: How to do impactful research in artificialintelligencefor chemistry and materials science:https://doi.org/10.1039/D4FD00153B
[2]
Sample EfficiencyMatters: A Benchmark for Practical Molecular Optimization:https://proceedings.neurips.cc/paper_files/paper/2022/file/8644353f7d307baaf29bc1e56fe8e0ec-Paper-Datasets_and_Benchmarks.pdf
[3]
Du, Y., Jamasb, A.R., Guo, J. et al. Machine learning-aided generative molecular design. Nat Mach Intell6, 589–604 (2024): https://doi.org/10.1038/s42256-024-00843-5
[4]
A. Duval, S. V. Mathis, C. K. Joshi, V. Schmidt, S. Miret, F. D. Malliaros, T. Cohen, P. Lio, Y. Bengio and M. Bronstein, arXiv, 2023, preprint,arXiv https://arxiv.org/abs/2312.07511
[5]
M. C. Ramos, C. J. Collison and A. D. White, arXiv, 2024, preprint,arXiv https://arxiv.org/abs/2407.01603
-
人工智能
+关注
关注
1791文章
46952浏览量
237810 -
机器学习
+关注
关注
66文章
8386浏览量
132473 -
深度学习
+关注
关注
73文章
5495浏览量
121044
发布评论请先 登录
相关推荐
名单公布!【书籍评测活动NO.44】AI for Science:人工智能驱动科学创新
《AI for Science:人工智能驱动科学创新》第一章人工智能驱动的科学创新学习心得
AI for Science:人工智能驱动科学创新》第4章-AI与生命科学读后感
从入门到研究,人工智能领域最值得一读的10本资料(附下载)
中科曙光异构智能算力技术加速计算材料科学创新变革
科研进阶!EPR在材料科学中的应用交流会即将开幕

如何在化学和材料科学领域开展有影响力的人工智能研究?(二)

如何在化学和材料科学领域开展有影响力的人工智能研究?(三)





 工商网监
工商网监
评论