 如何同时使用Nucleus与TensorFlow解决基因组学领域的机器学习问题
如何同时使用Nucleus与TensorFlow解决基因组学领域的机器学习问题
简介
在本文中,我们将 DNA 测序纠错表述为多级分类问题,并提出两种深度学习解决方案。第一种方法是在单次读取中纠错,而第二种方法(如图 1 所示)则通过多次读取来达成共识,以预测正确的 DNA 序列。我们的Colab 笔记教程使用Nucleus和TensorFlow库实现第二种方法。本文旨在向您展示如何同时使用 Nucleus 与 TensorFlow 解决基因组学领域的机器学习问题。

问题概览
尽管 DNA 测序日渐快捷和便宜,其过程仍容易出错。使用Illumina等公司开发的新一代测序 (NGS) 技术处理原始数据时,错误率约为 1%。第三代技术,例如Pacific BioSciences(PacBio) 公司开发的技术,正日益普及,其错误率约为 15%。测序错误可分为替换、插入和缺失,后两者通常称为 indel。所有这些错误均不利于下游的分析步骤,例如变异检测和基因组组装。
如要获取较高质量的数据集,一个简单的方法是舍弃可能包含错误的数据,丢弃全部读取内容或去除低质量区域皆可。该方法并非理想之选,因为这会导致最终的数据集会变小。此外,某些序列上下文本来就有较高的错误率,进而导致采样出现偏差。因此,大量研究都侧重于开发更成熟的纠错方法。大多数已开发的方法均可归类为以下两组之一:
对单次读取进行操作的方法,旨在确定正确的读取序列
对多次读取进行操作的方法,以共识为基础,旨在确定正确的基础 DNA 序列
深度学习概览
本文中阐述的两种方法均使用深度神经网络,学习将输入映射至输出的函数。神经网络由若干层线性与非线性运算构成,而这些运算会依次应用至输入。神经网络已成功应用于包括图像分类和自然语言翻译在内的多个问题领域。最近,神经网络也被用于解决基因组学问题,例如蛋白质结构预测和变异检测。
方法
Nucleus
我们的实现需要Nucleus,这是由 Google Brain 的 Genomics 团队开发的内容库,用于处理基因组学数据。Nucleus 使用专门的 reader 对象与 writer 对象,可轻松读取、写入和分析常见基因组文件格式(如 BAM、FASTA 和 VCF)中的数据。Nucleus 让我们能够:
针对指定基因组区域中的所有变异查询 VCF 文件
针对映射至指定基因组范围的所有读取内容查询 BAM 文件
针对从指定位置开始的参考序列查询 FASTA 文件
我们还能使用 Nucleus 将数据写入TFRecords,这种二进制文件格式由协议缓冲区消息构成,可由 TensorFlow 轻松读取。读取 TFRecords 文件后,我们会使用Estimator API训练和评估卷积神经网络。
数据
以下是我们在实现过程中所使用的文件列表。所有数据均公开提供,且此 教程包含下载链接与说明。
NA12878_sliced.bam — 从 20 号染色体(位置 10,000,000–10,100,000)获得的 Illumina HiSeq 读取内容,降采样至 30x 的覆盖度
NA12878_sliced.bam.bai — NA12878_sliced.bam 的索引
NA12878_calls.vcf.gz — 瓶中基因组 NA12878 变异的真值集合
NA12878_calls.vcf.gz.tbi — NA12878_calls.vcf.gz 的索引
hs37d5.fa.gz — hs37d5 的参考基因组
hs37d5.fa.gz.fai 和 hs37d5.fa.gz.gzi — hs37d5.fa.gz 的索引文件
注:教程 链接
https://colab.research.google.com/github/google/nucleus/blob/master/nucleus/examples/dna_sequencing_error_correction.ipynb
网络架构
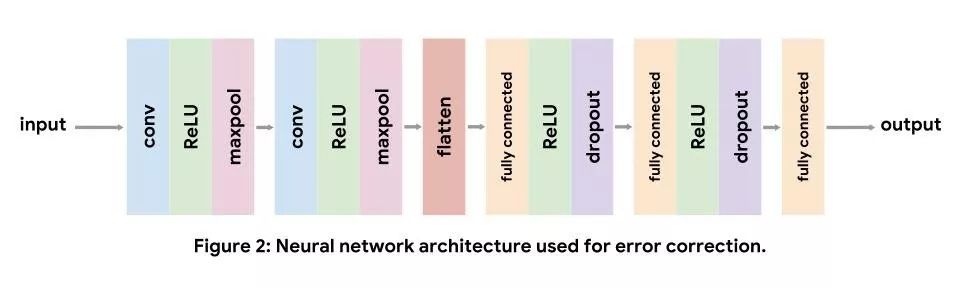
卷积神经网络通常用于处理计算机视觉任务,但也非常适用于基因组学。每个卷积层都会反复将学习后的过滤器应用于输入数据。在网络中早期出现的卷积过滤器会学习识别输入数据的低级特征(如图像中的边缘及色彩梯度),而后期出现的过滤器则会学习识别更复杂的低级特征组合。对于 DNA 序列输入,低级卷积过滤器会充当 motif 检测器,这类似于序列标识图的位置权重矩阵。
在实现过程中,我们使用的标准卷积架构依次由两个卷积层及三个全连接层组成。我们使用非线性 ReLU 层提升模型的表现能力。当卷积层减少输入量后,我们会进行最大池化,并会在全连接层充当正则化矩阵后退出此过程。请注意,在得到最终的全连接层后,我们不会加入 softmax 层,因为我们使用的损失函数是在内部应用 softmax。如需了解每层的详情,请参阅此教程。
注:教程 链接
https://colab.research.google.com/github/google/nucleus/blob/master/nucleus/examples/dna_sequencing_error_correction.ipynb
方法 1:单次读取的纠错
为了纠正序列读取中的错误,我们使用深度学习来训练神经网络,以解决一个较为普遍的问题:填充 DNA 序列中缺失的碱基。此方法旨在开发一种可理解 DNA 序列语法的模型。若仅靠真实序列的语法,我们可能无法获取充足的信息来开发可用于生产环境的解决方案。尽管如此,这依然是一个简单明了的示例应用。
出于指导目的,我们通过以下方法简化此问题:
仅考虑存在替换错误的区域,并忽略 indel 错误
仅考虑未存在已知变异的区域
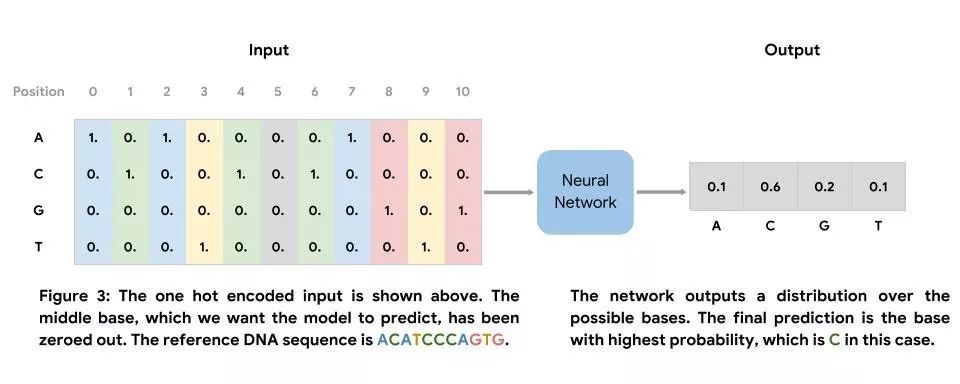
我们可以在参考基因组的区域中训练该神经网络。此网络的输入是定长的 DNA 序列,其核心是我们希望预测的碱基。此网络的输出是可能出现的碱基分布,且最终预测结果为可能性最高的碱基。我们使用在参考基因组中观测到的碱基产生标签集。由于我们仅使用映射至未存在已知真值变异之区域的读取内容,因此可以将参考基因组中存在的碱基明确标记为标签。
我们将参考基因组分割成非重叠的定长片段,以产生输入序列。在训练、评估和测试时,我们将参考序列中的一个碱基置零,以模拟缺失的碱基,如图 3 所示(位置 5)。除了使用参考基因组来模拟缺失的数据之外,我们还可将此类模型应用于序列读取的数据,特别是质量评分低于阈值的碱基。
方法 2:基于共识的纠错
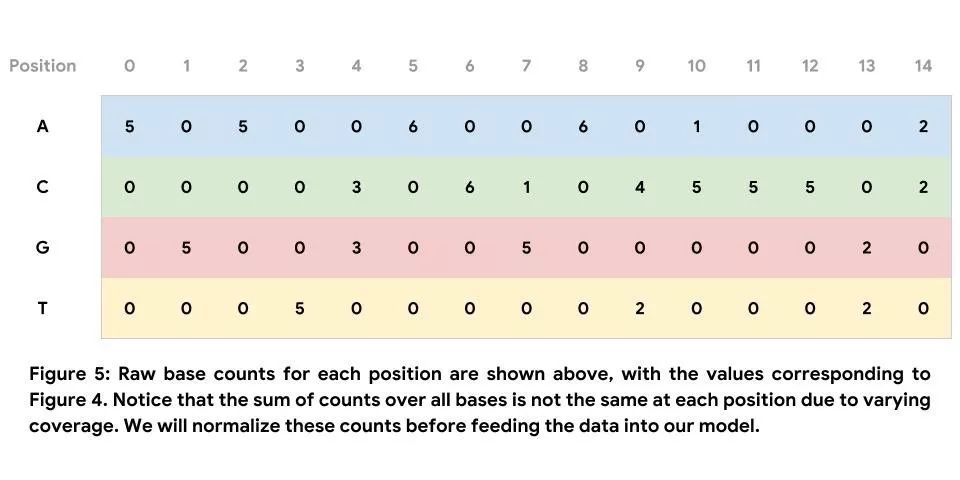
纠错的最终目的是确定基础 DNA 序列,而非为了纠正单次读取的错误。在本部分,我们通过汇总序列堆叠来使用多次读取达成的共识。如此一来,无需纠正单次读取的中间步骤即可直接确定 DNA 序列。有关序列堆叠的示例如下方图 4 所示。请注意,下图仅展示了此窗口中存在的读取部分。

出于指导目的,我们再次通过以下方法简化此问题:
仅考虑存在替换错误的区域,并忽略 indel 错误
仅考虑未存在已知变异的区域
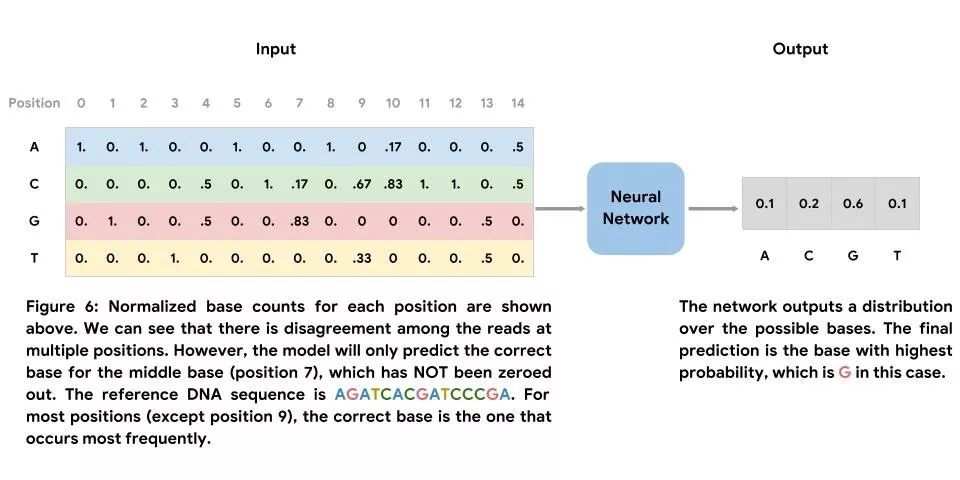
与第一种方法不同,我们并未在参考基因组中训练此模型。相反,我们的训练数据来自所映射的 Illumina HiSeq 读取内容。此网络的输入是在所映射的读取内容中观测到的标准化碱基数矩阵,其核心是我们希望预测的正确碱基的位置。Clairvoyante(一种用于变异检测的神经网络)的作者以及Jason Chin 的示例方法中皆使用了类似的特征化方式。此网络的输出是可能出现的碱基分布,且最终预测结果为可能性最高的碱基。与第一种方法类似,我们使用在参考基因组中观测到的碱基来产生标签集。我们将包含错误(在堆叠中至少有一次读取与中心位置中的参考序列不符)的示例及未包含错误(堆叠中的所有读取均与中心位置中的参考序列相符)的示例结合使用。
结论
此随附教程演示了本文所述的第二种方法。尽管我们分析的示例较为简单,不适合在生产环境中部署,但我们希望它们能帮助开发者学会高效利用 Nucleus 和深度学习解决基因组学领域的问题。
-
机器学习
+关注
关注
66文章
8453浏览量
133164 -
深度学习
+关注
关注
73文章
5521浏览量
121657 -
tensorflow
+关注
关注
13文章
329浏览量
60663
原文标题:使用 Nucleus 与 TensorFlow 进行 DNA 测序纠错
文章出处:【微信号:tensorflowers,微信公众号:Tensorflowers】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
基于大数据的食品组学精进安全食品
全基因组数据CNV分析简介 精选资料分享
全基因组测序的优势 精选资料分享
使用Arm服务器减少基因组学的时间和成本
利用基因组学和人工智能延年益寿将在未来可期
AI加速推动医疗个体化转型 基因组学将有望成为未来发展主流
为什么基因组是决定一个人特征的独特遗传密码
基于计算分析的高可用弹性宏基因组学计算平台
北鲲云超算平台对于基因组学研究能够提供哪些帮助?
NVIDIA Clara Parabricks助力基因组学和药物研究
微流控芯片技术在单细胞基因组学研究中的应用






 工商网监
工商网监
评论