 机器学习:快速精确预测电子结构问题
机器学习:快速精确预测电子结构问题

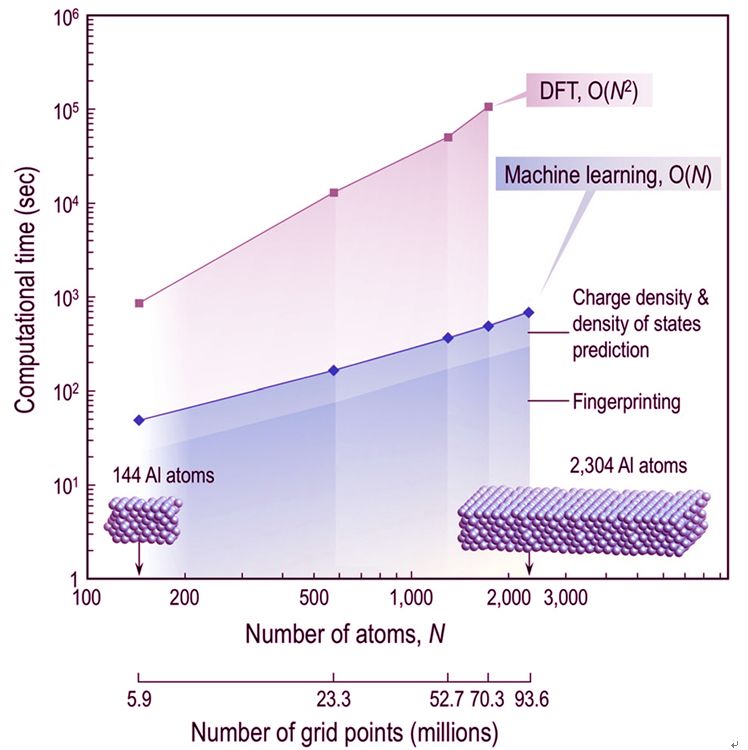
基于求解密度泛函理论(DFT)Kohn-Sham(KS)方程的模拟,已成为现代材料学和化学研究和开发组合过程的重要组成部分。尽管KS方程具有很强的普适性,但由于求解计算量很大,常规DFT计算一般只限于几百个原子。
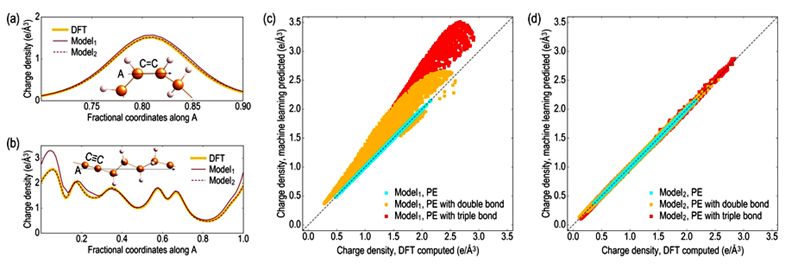
来自佐治亚理工学院的RampiRamprasad领导的团队,报道了一种基于机器学习的方法,可以不直接求解KS方程而有效预测电子结构。该方法利用新的旋转不变表示,将格点周围的原子环境映射到该格点处的电子密度和局部态密度,并使用预先计算得到的带有几百万的格点信息的DFT结果来训练的神经网络来获得该映射。上述方法可以精确模拟实际求解KS方程的结果,但是速度快几个数量级。此外,由于该方法的计算量与系统尺寸严格成线性关系,因而有望用于大型体系的电子结构预测。
该文近期发表于Computational Materials5:22(2019)
Solving the electronic structure problem with machine learning
Anand Chandrasekaran, Deepak Kamal, Rohit Batra, Chiho Kim, Lihua Chen & Rampi Ramprasad
Simulations based on solving the Kohn-Sham (KS) equation of density functional theory (DFT) have become a vital component of modern materials and chemical sciences research and development portfolios. Despite its versatility, routine DFT calculations are usually limited to a few hundred atoms due to the computational bottleneck posed by the KS equation. Here we introduce a machine-learning-based scheme to efficiently assimilate the function of the KS equation, and by-pass it to directly, rapidly, and accurately predict the electronic structure of a material or a molecule, given just its atomic configuration. A new rotationally invariant representation is utilized to map the atomic environment around a grid-point to the electron density and local density of states at that grid-point. This mapping is learned using a neural network trained on previously generated reference DFT results at millions of grid-points. The proposed paradigm allows for the high-fidelity emulation of KS DFT, but orders of magnitude faster than the direct solution. Moreover, the machine learning prediction scheme is strictly linear-scaling with system size.
-
电子
+关注
关注
32文章
2044浏览量
94060 -
机器学习
+关注
关注
67文章
8570浏览量
137390
原文标题:npj: 机器学习—快速精确预测电子结构问题
文章出处:【微信号:zhishexueshuquan,微信公众号:知社学术圈】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
瑞萨电子AI解决方案助力优化电子配送机器人开发

学习单片机快速方法
从数据到模型:如何预测细节距键合的剪切力?
机器学习和深度学习中需避免的 7 个常见错误与局限性

《AI机器人控制进阶教程(入门版)》阅读指引

提高条件分支指令预测正确率的方法
基于全局预测历史的gshare分支预测器的实现细节
机器视觉检测PIN针
贸泽电子2025边缘AI与机器学习技术创新论坛回顾(上)
FPGA在机器学习中的具体应用
机器学习异常检测实战:用Isolation Forest快速构建无标签异常检测系统

明远智睿SSD2351开发板:语音机器人领域的变革力量
贸泽电子2025技术创新论坛探讨“边缘AI与机器学习”新纪元




评论