 电子发烧友App
电子发烧友App


自诞生以来,GPU就被广泛应用于高性能科学计算领域。GPU的大规模并行计算能力帮助包括分子模拟在内的许多传统科学计算应用实现了爆炸式提速。分子模拟是一种利用计算机在微观尺度下研究材料、蛋白质、药物等分子行为的技术。由于其模拟流程中包含许多高计算量的模块,分子模拟程序非常适合通过GPU来加速。经过十几年的发展迭代,目前大部分常用的分子模拟软件都对GPU硬件进行了不同程度的适配。本文将对分子模拟中关键的近程力计算模块在三款不同模拟软件中的GPU移植案例展开深度剖析,希望能以小窥大,探究科学计算应用在GPU上优化的关键点。
在本系列的上篇《分子模拟:打开微观世界的钥匙(上)—— 起源、发展与现状》,我们介绍了分子模拟技术的起源、发展与现状。分子模拟技术可以通过一个体系的化学组成和其所处的环境来预测其物理/化学以及动力学性质,是科学计算领域的一个重要组成部分。而作为一项历史悠久的仿真技术,传统的分子模拟程序通常都是通过多线程的方法在CPU上进行模拟。但在GPU横空出世以后,人们惊喜地发现,在GPU的超高并行效率的协助下,分子模拟的速度和模拟体系的尺寸都可以得到数量级的提升。因此,目前的大部分主流分子模拟应用中都加入了GPU版本供用户使用。然而,仔细研究后可以发现,各大应用在GPU上的适配方法差异很大。本文将以分子动力学计算中的近程力计算模块(short-range force)为例,拆解几大主流应用中的GPU算法细节。本文中提到的GPU术语皆以CUDA体系为标准,包括:thread(线程), warp(线程束), shared memory(共享内存), host(主设备), device(计算设备)等。
1
分子动力学近程作用力计算模块
本系列上篇曾经介绍过,分子动力学模拟中体系力场的计算可以被大致分为两大部分:成键作用力(bonded interactions)和非成键作用力(nonbonded interactions)。顾名思义,成键作用力计算的是互相形成化学键的原子之间的作用力,而非成键作用力则是体系中所有没有形成化学键的原子间的相互作用力。由于非成键作用力的计算需要遍历体系中所有可能的原子对,其复杂度达到了 O(N2) (此处N为体系中的总原子数)。因此,非成键作用力的计算是分子动力学模拟中最耗时的部分。考虑到该作用力随距离的增加而快速衰减的特性,我们可以通过引入截断距离(cutoff distance)的概念来简化计算。我们将小于截断距离的部分作用力称为近程作用力,而大于截断距离的部分则称远程作用力。
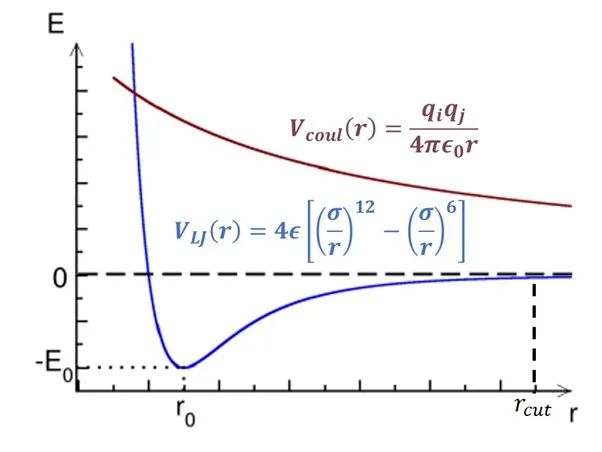
图1:非成键作用势随距离变化简图。
横轴为距离,纵轴为势能。
如图1所示,范德华势(VLJ, 色散作用)和库伦势(Vcoul, 电荷作用)都随距离r的增加而递减。其中范德华势在截断距离(rcut)处基本衰减到零,因此大部分情况下这部分计算只需要考虑截断距离内的原子对。但库伦势衰减速度较慢,我们无法直接忽略其长程(>rcut)部分。库伦势的长程部分可以通过目前最先进的PME(particle-mesh Ewald)方法[1]计算,该算法的时间复杂度仅为O(NlogN)。由此可见,近程作用力的计算(O(N2))是模拟体系力场算法中最消耗计算资源的部分。如图2所示,非成键作用力中的近程力计算开销(SR+NEIGH)在总开销中的占比高达~80%。因此,近程力计算模块的加速是提升模拟速度的关键所在,该模块也成为了最早被移植到GPU上的计算单元,目前各大主流模拟软件中都有较为成熟的GPU近程力计算版本。后面几个小节中,笔者将讨论近程力计算模块在CPU上和不同软件的GPU版本中的算法差异。
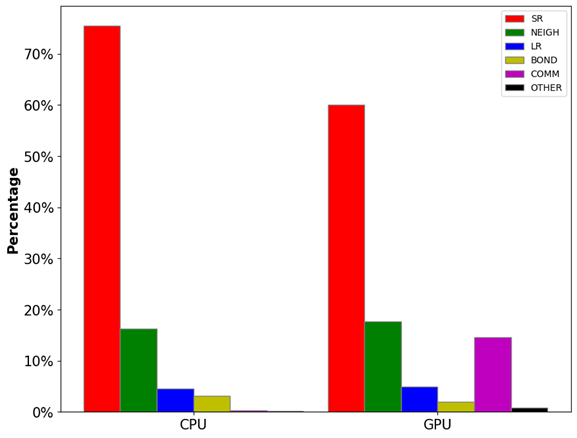
图2:LAMMPS分子动力学模拟中各模块的计算开销占比。其中,SR为近程力计算开销,NEIGH为邻近列表构建开销,LR为长程力计算开销,BOND为成键作用力计算开销,COMM为通讯开销,OTHER为其他开销。
测试体系:视紫红质(Rhodopsin)蛋白质体系,96,000原子,LAMMPS Benchmarks
2
近程力CPU算法
对于一个包含N个原子的体系,计算近程作用力最直接的方法就是双循环遍历所有可能的原子对,并通过距离和力场参数计算部分作用力,最后对所有结果进行归约操作。很明显,即使在引入截断距离以后,此种暴力求解方法的时间复杂度仍为O(N2)。因此,我们需要一些技巧来加速近程力计算模块,而neighbor list(邻近列表)方法就是一种最常用的技巧。neighbor list方法为体系内的每一个原子都构建一张列表,其中存储着在三维空间中与其临近(距离小于截断距离)的其他原子的编号。当这些列表构建完毕以后,在计算近程力时,我们只需要遍历每个原子与其neighbor list中的其他原子的作用力即可,因为截断距离外的作用力可以忽略不计。如此一来,近程力计算的复杂度就被减小到了O(N)。
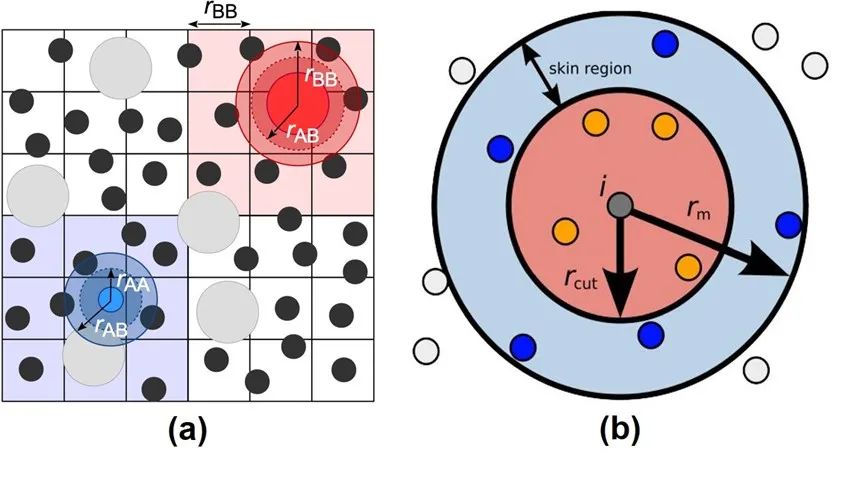
图3:Verlet Neighbor List方法示意图。
来源:Neighbor lists — HOOMD-blue 2.5.2 documentation
然而,构建neighbor list的时间是无法被忽略的。同时,由于在模拟过程中原子位置会持续发生变化,这些列表需要频繁地被重建。于是,人们提出了一种可以将neighbor list重复使用的技巧: Verlet neighbor list方法[2]。考虑到原子运动的连续性以及分子动力学模拟中通常步长很短等特性,原子的坐标在若干步模拟过后通常不会改变太大。因此,如图3(a)所示,我们可以把模拟体系划分成若干网格, 将每一个原子归到格子里, 然后只需要搜寻原子当前以及周围总共27个(假设3D体系)网格中的其他原子即可。另外,为了记录原子进入或离开neighbor list范围的情况,我们还可以在截断范围以外设置一个缓冲区域(skin region), 如图3(b)所示。假如监测到有大量原子离开缓冲区,则需立刻重构Verlet list;否则,可以按一定的频率(一般每20到40步模拟)来更新list。如此便可以做到Verlet list的复用,节省大量计算开销。图4展示了CPU中近程力计算的两大模块,neighbor list build (a)和粒子间作用力计算(b)的简单计算流程。


图4:近程力CPU计算模块:
(a) neighbor list 构建, (b) 作用力计算。
3
近程力LAMMPS-GPU算法
本文后面的篇幅中,我将介绍三款分子动力学应用中的近程力GPU算法。首先要介绍的是LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)[3]。LAMMPS 是一款由美国桑迪亚国家实验室(Sandia National Lab)开发的开源分子动力学模拟程序包。该程序于1995年第一次发布,距今已有将近30年的历史。
LAMMPS的GPU包中对近程力模块的移植相对较为直接,并没有对数据结构进行大幅改变,只是在最新的版本中加入了更多warp-level指令的使用。首先,LAMMPS-GPU的粒子间作用力的计算基本与CPU版本(见图4(b))完全一致。简单来说,即GPU中每一个thread负责一个原子,循环计算该原子与其neighbor list内的原子的相互作用力,再将结果写入到储存作用力的数组中。而在neighbor list构建模块中,LAMMPS的GPU版本与CPU版本最大不同是通过warp shuffle的指令来减少从host到device的数据读取。具体来说,该算法分为以下步骤:

图5:LAMMPS-GPU neighbor list build模块计算流程。
得益于GPU的高度并行能力以及warp 内部的数据共享能力,LAMMPS的GPU版本确实比CPU提升了不少速度。然而,这种直接的GPU移植方式还是存在着许多的缺陷,包括:
01
没有改变传统neighbor list的结构,LAMMPS的neighbor list 数据结构如图6所示,每个粒子都有一个自己的neighbor list,导致有很多的neighbors被重复储存。使用这样的数据结构会大幅增加host-device间的数据传输压力。
02
网格是通过空间坐标均匀划分的,当处理非均质体系时各warp需要处理的粒子数差异较大,易造成线程分歧(thread divergence)。
03
计算粒子间作用力时没有利用好shared memory/warp shuffle功能来减少从host到device的数据传输。
由此,我们可以看到,想要更好地激发GPU的潜力,程序还需要对底层的数据结构和运算逻辑进行进一步优化。
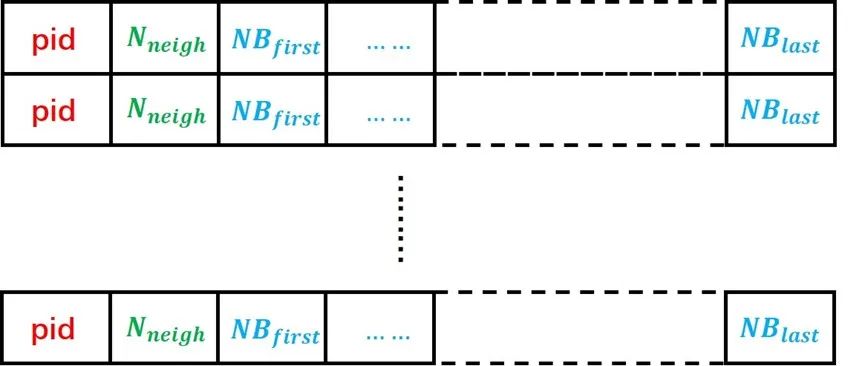
图6:LAMMPS中的neighbor list数据结构。每个原子对应其中的一行数据,pid为原子编号,Nneigh为该原子的neighbor总数,NBfirst至NBlast为从第一个neighbor到最后一个neighbor的编号。
4
近程力GROMACS-GPU算法
GROMACS(GROningen MAchine for Chemical Simulations)[4]是一款由荷兰格罗宁根大学团队开发的模拟程序,该软件于1991年首次发布,目前已经成为全球使用人数最多的分子动力学软件之一。GROMACS从2013年的4.5版本开始支持GPU,经过多次迭代优化之后,目前的最新版本已经可以在GPU上完成全模拟流程。
与LAMMPS-GPU不同的是,GROMACS的开发团队意识到了传统的neighbor list结构不适合GPU的SIMT(单指令多线程)计算特性。因此,他们提出了一种基于cluster(聚类)结构的新型近程力算法[5]。受到矩阵乘法运算的启发,该方法通过将体系中的粒子分为N x M大小的cluster来提升GPU的计算效率。
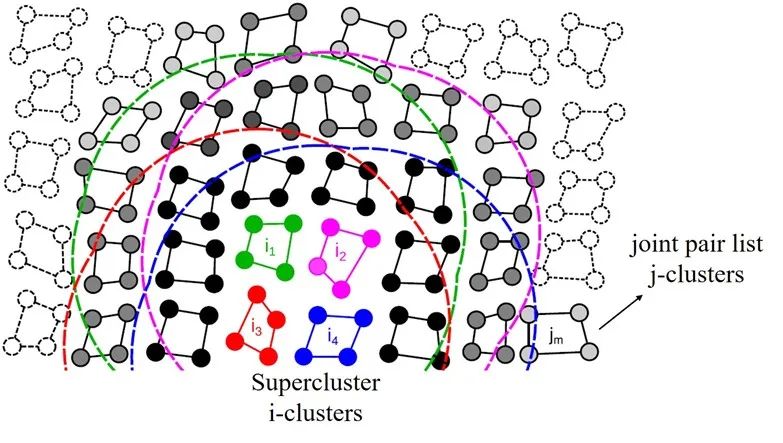
图7:GROMACS-GPU算法中的super-cluster结构。图中的super-cluster包含i1-i4 四个i-cluster,这些i-cluster的neighbors被整合成了一个joint pair list, 即图中黑色和灰色的j-cluster合集。
来源:Páll etc. “Heterogeneous parallelization and accelerationof molecular dynamics simulations in GROMACS”, J. Chem. Phys. 153, 134110 (2020)
以图7为例,图中的体系被分为包含N=4个粒子的i-cluster和包含M=4个粒子的j-cluster。不同于以单个粒子为计算单元的传统neighbor list算法,此新方法以粒子的聚类为单位来构建neighbor list。具体来说,在搜索neighbor的过程中,程序会遍历所有的i-cluster,并找到在其截断距离范围内的j-cluster加入其neighbor list。如此一来,在计算作用力的时候就可以每次同时载入M 个粒子的信息,从而减少对neighbor数据的重复读取操作。尽管这种做法会无可避免的增加一些无效计算(并不是cluster中的每个粒子都在目标粒子的截断范围内),但是对于这样一个瓶颈在内存操作的程序来说,使用该方法的收益还是远远大于损失。
在实际应用中,这种聚类临近列表(cluster neighbor list)方法可以进行进一步的优化来占满目标硬件的执行宽度(execution width)。以Nvidia的GPU产品为例,N卡以warp为计算单元,每个warp中包含32个thread,因此其执行宽度为32。我们可以通过将cluster的大小设置为N=8,M=4 来达到最高计算效率。同时,还可以将4个i-cluster合并成一个super-cluster(见图7中的i1-i4), 并将它们的neighbor list整合成一个联合成对列表(joint pair list)。如此操作的好处在于可以充分利用GPU中较为快速的shared memory来进一步减少host到device端的数据传输。在每一次计算作用力时,程序会先将super-cluster中所有粒子的数据读取进来并储存在shared memory中,这样就避免了在后续的计算过程中进行缓慢的显存读取(global memory load)操作。同理,在执行宽度为64的AMD GPU上,需要设置N=M=8 ; 而在英特尔硬件上则需将cluster的大小设置为N=4,M=2 。
通过对底层数据结构的改进优化,GROMACS成功提升了GPU计算资源的利用效率。与传统的neighbor list结构相比,cluster pair list的使用可以显著减少同一个neighbor的重复储存,在数据复用和内存使用等方面做到了很好的优化。同时,相对均匀的计算分配也可以大幅缩短thread的闲置和等待时间。尽管增加了一定的计算量,但是在这个内存瓶颈的程序中,这部分增加的计算时间可以被隐藏在内存操作延迟中,从而不对整体性能产生很大影响。因此,笔者认为GROMACS-GPU的近程力计算模块是一个很成功的GPU移植和优化案例。
5
近程力OpenMM-GPU算法
接下来要介绍的OpenMM[6]是一款主打易用性、拓展性和高性能的新型模拟引擎。该软件的开发团队主要来自斯坦福大学,他们设计这款软件的初衷就是想要解决传统分子模拟软件难上手、源码修改困难以及在GPU上性能不佳等问题。经过近十年的努力,OpenMM也确实越来越受学者们的欢迎。一方面,OpenMM的Python 和C++ API都做的非常完善,用户可以自由添加新功能或者修改现有功能。另一方面,OpenMM的GPU算法先进,加速效果非常好。因此,许多新的研究项目都选择其作为模拟引擎,包括著名的蛋白质结构预测模型Alpha Fold[7]。
为了提升近程力计算在GPU上的性能,OpenMM同样在neighbor list的数据结构上进行了优化。其整体的优化思路与GROMACS比较类似,也是通过将多个粒子集合成atom block(原子块)的方式来构建neighbor list,以减少neighbors的重复储存和数据传输。两种方法的最大区别在于每个集群中粒子数量的选择。OpenMM的atom block中包含32或64个原子,而GROMACS中的cluster则只有4或8个粒子。增加集群的尺寸可以减少neighbor list搜索和构建的开销,因为这部分计算的耗时是由体系中的集群总数决定的。但同时,大集群也意味着在作用力计算模块中会引入更多的无效计算,增加计算资源的浪费。因此,集群尺寸的选取实际上是近程力计算中两大模块之间的平衡取舍问题。OpenMM团队经过验证后发现较大的集群在现代GPU架构中可以更为高效。同时,他们还使用了一些技巧来降低增大集群尺寸后引入的负面效应。比如,在构建neighbor list的时候增加一个细致搜索的步骤。
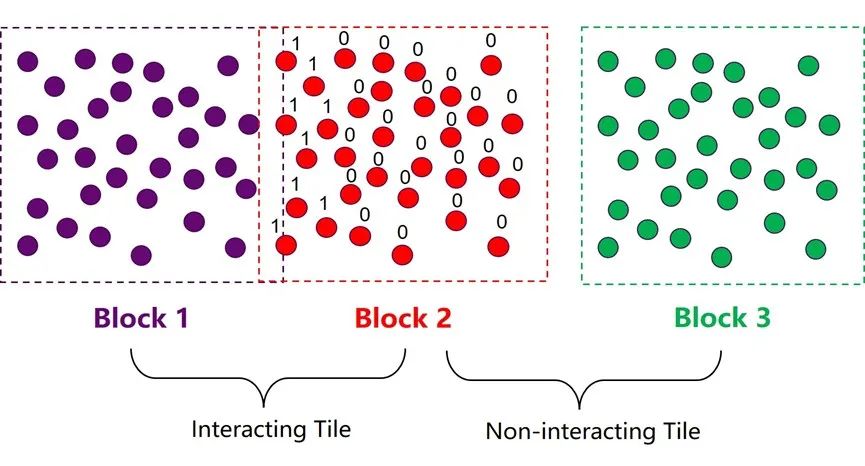
图8:OpenMM-GPU算法中的atom block 和tile结构。
如图8所示,在第一步粗略搜索的过程中,程序会将边界有交集的两个atom block 标记为相互作用集群对(interacting tile);边界没有交集的atom block对则被标记为互不作用集群对 (non-interacting tile)。这一步搜索结束以后,程序会马上进入下一步细致搜索阶段,这一步的目的是构建一个32/64位的mask,对block 2中的所有粒子进行判定,假如一个粒子与block 1中的任意粒子有相互作用,则将该位设为1,反之则设为0。这个mask中储存的信息可以有效规避后续作用力模块中的大部分冗余计算。OpenMM中这种类似的小trick还有很多,正是因为他们对GPU更深的理解和对细节的注重,使得OpenMM在GPU上的加速效果达到了业界最顶尖的水平。
结语
本文介绍了三款常用的分子动力学模拟应用中的GPU加速算法,分别代表三种不同程度的GPU适配思路。其中,LAMMPS的GPU版本基本直接沿用了CPU上的算法,只是利用GPU的高度并行能力对其中的计算密集部分进行加速。而GROMACS和OpenMM则将CPU版本中的底层数据结构和计算逻辑进行了大刀阔斧地改造,以更好地发挥GPU的硬件特性。另外,OpenMM通过一些算法细节上的巧妙设计进一步提升了程序在GPU上的运行效率。
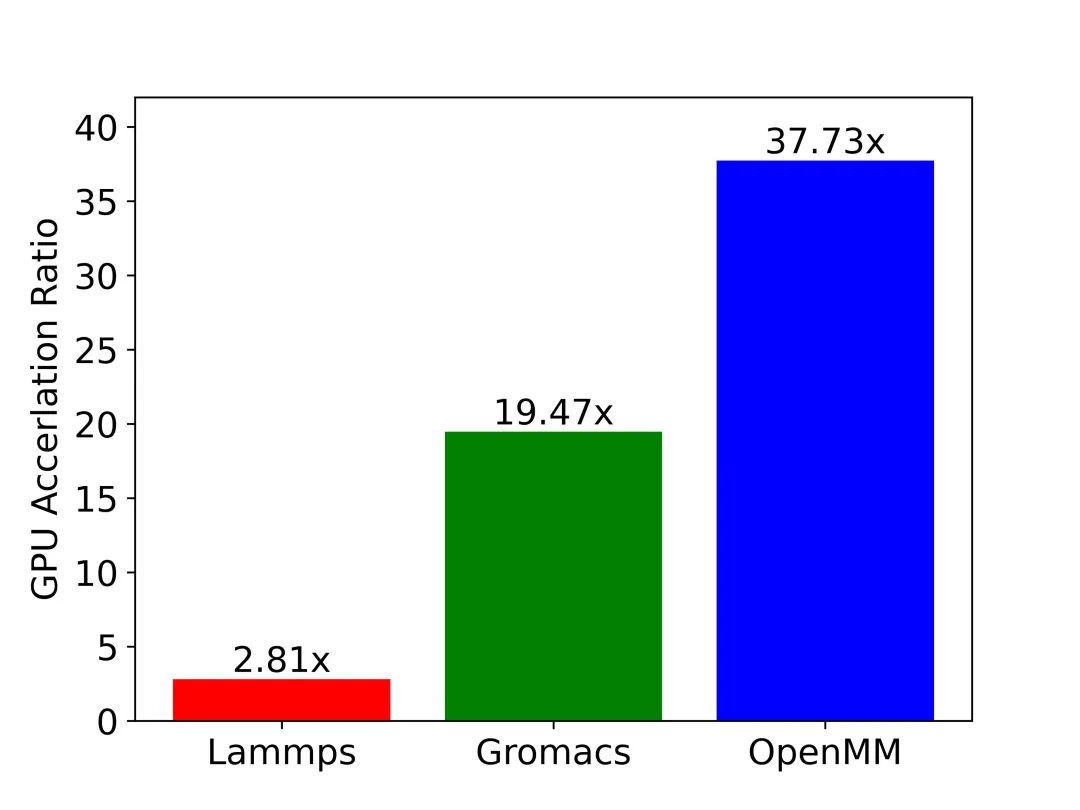
图9:三大应用GPU加速效果一览。图中纵轴为GPU相对CPU的加速比。测试用例:包含45,000原子的SPC/E模型水体系,通过沐曦MetaMol分子模拟工作流软件实现相同体系在不同应用上的模拟。
测试硬件:CPU: AMD Ryzen 7 3700X 8-Core Processor; GPU: NVIDIA GeForce RTX 3080。
图9中展示了LAMMPS、GROMACS和OpenMM在模拟SPC/E模型水体系时的GPU-CPU的加速比数据。测试结果与前文中的分析一致:在相同的硬件上模拟同一体系时,OpenMM的GPU加速效率要远高于GROMACS和LAMMPS。读到这里大家应该都有疑问:为什么LAMMPS不对数据结构进行优化呢?究其原因,以LAMMPS为代表的传统软件历史悠久,在设计之初并没有考虑到在不同平台、不同硬件上适配的需求,导致软件的移植工作量巨大,而修改底层数据结构更是牵一发而动全身,几乎难以完成。另一方面,像OpenMM这种近年来开发的新型应用则对软件的拓展性和灵活性非常重视。比如在OpenMM中,用户可以很便捷地通过插件的形式来添加新的计算平台或是新算法。因此,在各种硬件架构层出不穷,日新月异的今天,如何保证拓展性和灵活性需要成为我们开发科学计算软件时必须考虑的问题。
个人认为这些软件的GPU移植和优化的实例比较具有研究价值,希望这篇文章可以给各位读者带来一些启发,拓宽一些思路。本文中的大部分内容都来自笔者自己对软件源码的研究和模拟数据的分析所得。文中提出的观点都来自我的个人理解,难免有所纰漏,欢迎大家批评指正。最后,希望通过进一步的优化和改进,我们可以让分子模拟在沐曦的通用GPU上达到更高的速度和精度,真正成为人类打开微观世界的钥匙。
编辑:黄飞






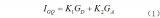
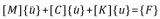







 工商网监
工商网监
评论