01 导读
在传统可充电锂离子电池(LIB)中,以碳酸亚乙酯(EC)为主的Li+初级溶剂化鞘(PSS)在Gr上能够形成独特的固体电解质界面(SEI),抑制溶剂的共嵌入,并避免Gr的结构坍塌。为了提高电池安全性,在EC基电解质中引入有机磷酸盐作为共溶剂,可以增加电解液的阻燃性。
然而,大多数有机磷酸盐表现出高的极性和强的给电子能力,可以与Li+配位。因此,磷酸盐分子进入PSS并部分取代EC分子(图1a)。通过这种方式,磷酸盐参与了初级SEI的形成,并破坏了原来的SEI。此外,配位磷酸盐倾向于与Li+共嵌入Gr晶格中。插层分子在低电位下的分解会使Gr颗粒破裂,并导致性能迅速下降。因此,在不干扰电极材料储锂性能的情况下构建阻燃电解质仍然具有挑战性。
02 成果背景
近日,JACS上发表了一篇题为“Noncoordinating Flame-Retardant Functional Electrolyte Solvents for Rechargeable Lithium-Ion Batteries”的文章,该文章提出了一种非配位阻燃共溶剂,能够提高电池安全性,同时避免对储锂过程的干扰。选用具有良好理化性能的高效阻燃剂六氟环三磷腈作为共溶剂制备功能电解质。
给电子能力低的非极性磷腈分子不能与Li+配位,因此被排除在初级溶剂化鞘外。在基于石墨负极的锂离子电池中,磷腈分子在充电过程中不会与锂离子共插进入石墨晶格中,有助于保持负极结构和界面的完整性,实现稳定循环。
03 关键创新
(1)六氟环三磷腈(HFP)作为一种非配位的阻燃共溶剂,不参与构建Li+溶剂化鞘,因此不会破坏原有的SEI结构。
(2)HFP不与Li+共插入石墨层间,因此避免了石墨负极结构和界面的破坏。
(3)HFP的高阻燃特性极大提高了电池安全性。
04 核心内容解读
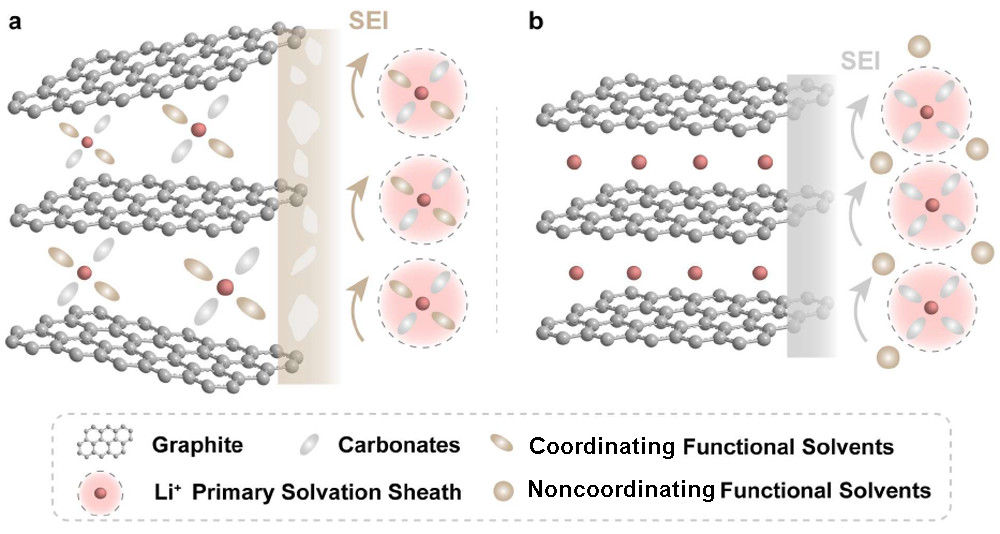
图1. 在(a)具有配位共溶剂的电解质和(b)具有非配位共溶剂的电解质中,Li+嵌入Gr的示意图。
本工作提出了一种新的非配位共溶剂——六氟环三磷腈(HCP),它可以提高电解质阻燃性,但对Li+溶剂化结构的影响最小(图1b)。HCP是一种全氟六元芳族磷腈分子,作为阻燃共溶剂,被引入传统的碳酸盐电解质E1(0.8 M LiPF6 in EC/碳酸二乙酯(DEC),1:1.5 m/m),获得功能性电解质E2(0.8 M LiPF6 in EC/DEC/HCP,11 mm)。通过将经典的磷酸盐TMP加入E1来制备电解质E3(0.8 M LiPF6 in EC/DEC/TMP,11 mm),并作为对照组。E1和E3可点燃并剧烈燃烧。相反,E2不能被点燃。
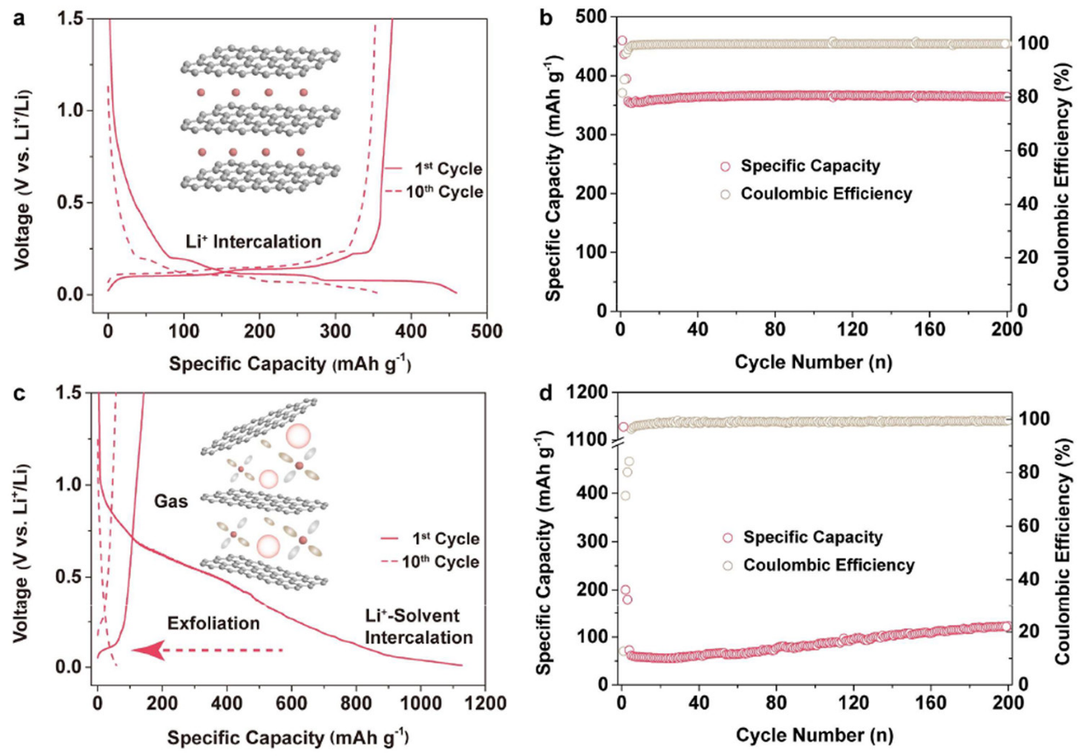
图2. 使用(a, b)E2和(c, d)E3电解质的Li||Gr电池(a, c)恒流充放电曲线和(b, d)循环性能。
作为锂离子电池最常用的负极材料,Gr对电解质和SEI有着严格的要求。为了研究上述电解质的电化学相容性,对Li||Gr电池进行了测试。在初始放电过程中,Li|E1|Gr和Li|E2|Gr电池的比容量均约为460 mAh g-1,具有三个电压平台,分别对应于SEI形成和Li+嵌入Gr(图2a)。相比之下,Li|E3|Gr电池的比容量大于1000 mAh g-1,电压曲线表明发生了大量溶剂共嵌入和分解(图2c)。
Li|E1|Gr和Li|E2|Gr表现出稳定的循环性能,并在200次循环后,保持约100%的初始充电容量(图2b),而Li|E3|Gr容量迅速下降(图2d)。显然,将HCP引入碳酸盐电解质不会影响与Gr的相容性。
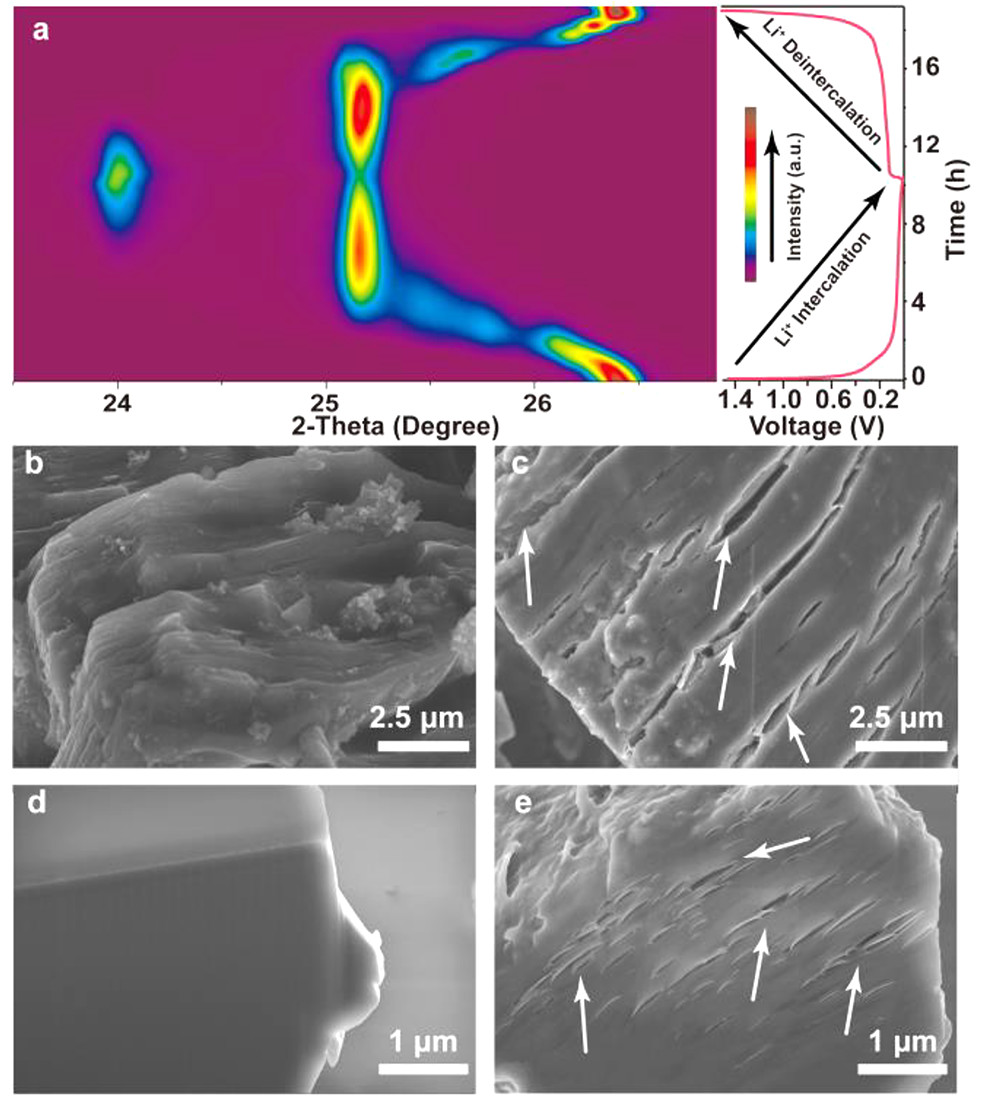
图3. Li|E2|Gr电池(a)第一次充放电循环(0.1C, 0.01-1.5 V)的原位XRD等高线图。在(左)E2和(右)E3电解质中第一次循环后,Gr的(b, c)SEM图和(d, e)FIB-SEM图。@ACS
接下来,研究了在不同荷电状态下Gr的X射线衍射(XRD)谱图。Li|E2|Gr电池充放电过程的原位XRD谱图(图3a)显示,Gr的(002)峰位置在Li+插层过程中由26.4°变为23.7°,放电过程中向相反方向对称移动,表明Li+插层具有高的可逆性。SEM图像显示,在E3中循环后的Gr颗粒明显膨胀,表面(图3c)和内部(图3e)均有明显裂纹。
相反,E2(图3b,d)中循环的Gr颗粒保持良好,没有可见裂纹。这些明显的裂纹和Gr在E3中的巨大径向膨胀(约10倍)可能是由于共插的溶剂分子分解导致层状结构剥落。
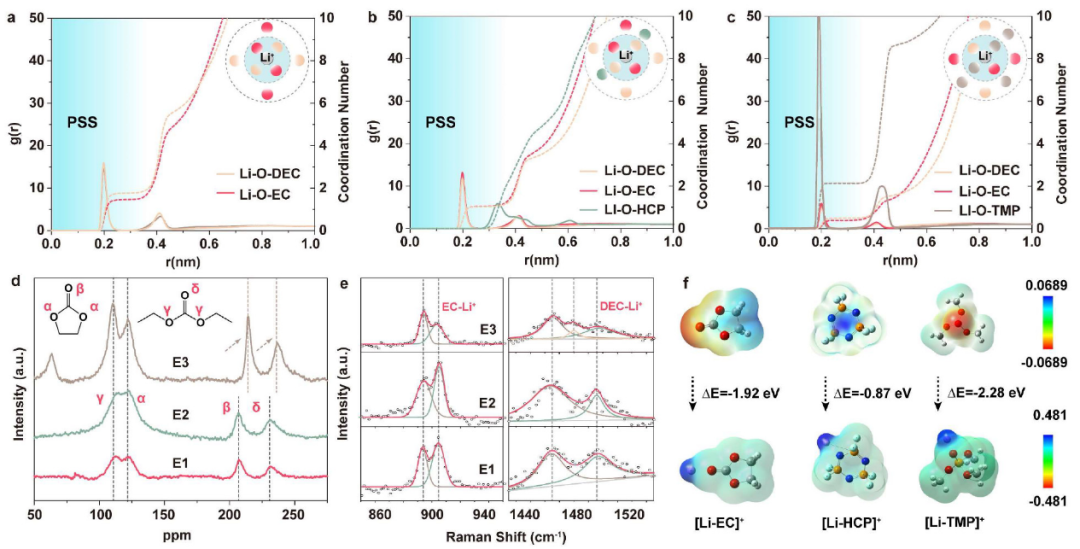
图4. (a-c)不同电解质的Li-O径向分布函数。所附原理图显示了相应的溶剂化结构。不同电解质的(d)17O核磁共振谱和(e)拉曼光谱。(f)溶剂和溶剂化结构的静电势分布。@ACS
接下来,通过分子动力学(MD)模拟来说明Li+在不同电解质中的溶剂化结构(图4a-c)。首先,对溶剂化层中与Li+相互作用的溶剂分子进行计数和平均。如图4c所示,E3的Li+ PSS中存在TMP,平均配位数为2.13,远远大于EC和DEC的配位数(分别为0.39和0.49)。相比之下,HCP几乎没有参与PSS(r<3 Å),E2中的平均配位数接近于0(图4b)。与E1的情况一样(图4a),由于HCP的非配位性,EC和DEC在E2的Li+ PSS中仍然占主导地位。
采用密度泛函理论(DFT)模拟计算溶剂和溶剂化结构的形成能和静电势(图4f)。相对于HCP, TMP和EC具有更强的表面负电荷,表现出更强的给电子能力和Li+配位能力。[Li+-溶剂]形成能可定义为
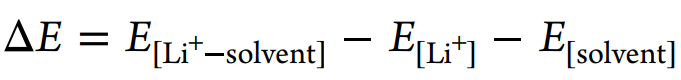
ΔE[Li+-EC](−1.92 eV)比ΔE[Li+-HCP](−0.87 eV)大得多,但小于ΔE[Li+TMP](−2.28 eV),说明这三种溶剂的配位能力为TMP>EC≫HCP, HCP不能在热力学上取代EC。
通过17O核磁共振光谱、拉曼光谱和电喷雾质谱(ESI-MS),进一步研究了电解质的溶剂化结构。如图4d所示,EC和DEC中有两组不等效的17O核,羰基17O(β和δ)位于200和250 ppm之间,醚17O(α和γ)位于75和150 ppm之间。E3中羰基17O核经历了下场位移,脱屏蔽效应增强,这可能与TMP参与Li+ PSS以及Li+与碳酸盐溶剂之间离子偶极相互作用减弱有关。
相反,E2中没有发现位移,说明HCP不影响[Li+-碳酸盐]的溶剂化结构。905 cm-1处的拉曼峰(图4e)代表[Li+-EC]键。相对于E1, E3中905 cm-1处的峰强度明显降低,而E2中无明显变化。在1486 cm-1处的[Li+-DEC]峰也可以观察到类似的结果。这表明HCP对碳酸盐的溶剂化状态影响较弱,证实了其非配位性。
05 成果启示
本工作提出了一种非配位的磷腈共溶剂,以提高电解质的阻燃性,同时避免干扰LIBs中的电荷转移和存储过程。与传统的有机磷酸盐不同,非配位HCP分子不参与Li+ PSS的形成,也不与Li+共插到负极结构中,因此不会导致负极-电解质界面退化或SEI不稳定。基于该新型电解质的LIB具有良好的循环性能。该新型非配位阻燃共溶剂与其他正极稳定剂结合,有望协同提高可充电电池的存储和安全性能,在未来的电化学系统中具有广阔的应用前景。
审核编辑:刘清