1
研究背景
室温钠硫电池因其理论能量密度高、成本低、资源丰富、环境友好等优点而受到人们的广泛关注。但它仍然面临以下挑战:(1)硫正极及其放电产物的低电导率,反应动力学缓慢,多硫化钠(NaPS)的穿梭效应。(2)Na枝晶的形成。由活性中心组成的极性纳米结构材料能有效吸附极性NaPS,并可作为电催化剂,加速NaPS转化为Na2S。
但是,低密度的催化活性位点和过量的金属负极会抵消Na-S电池的高能量密度优势。另外,在Na金属负极中,轻质多孔碳材料由于其优异的导电性和化学稳定性,有望抑制Na枝晶形成。然而,大多数碳骨架通常是疏钠的。因此,针对高能量密度Na-S电池体系,研究一种能同时调节阳离子和阴离子迁移行为的协同策略具有重要意义。
2
成果简介
近日,清华大学李亚栋院士和王定胜副教授联合中科院福建物质结构研究所温珍海教授在JACS上发表了题为“Single-Atom Yttrium Engineering Janus Electrode for Rechargeable Na–S Batteries”的论文。该论文采用金属-有机框架(MOF)制备了一个单原子杂化物,其中Y单原子被引入氮掺杂的四边形碳宿主(Y SAs/NC)中,该杂化物具有良好的亲钠和亲硫Janus性质,因此当用作钠-硫全电池的钠负极和硫正极的宿主时具有良好的电化学性能。
该Na-S全电池能够提供822 mAh g-1的高容量,并显示出优异的循环稳定性(在5 A g-1的下,循环超过1000圈后的容量保留率为97.5%)。3D打印电池和Na-S软包电池进一步验证了这种Na-S电池的实际应用潜力。
3
研究亮点
(1)理论计算表明,YN4/C可以降低Na2S的分解能垒,表现出较强的多硫化物(Na2S6)吸附能,并能够提供有效的亲钠位点,促进钠均匀成核。 (2)因此,将Y单原子引入到N掺杂碳多面体(Y SAs/NC)中,构建了Y SAs/NC-S||Y SAs/NC-Na全电池,该电池具有高容量、优异的倍率性能和循环稳定性。 (3)机理研究表明,原子Y可以电催化S8还原为Na2S,加速反应动力学,有效缓解“穿梭效应”,并在高电流密度下表现出稳定的循环性能。
4
图文导读
首先,通过DFT计算预测了YN4/C的电催化SRR动力学和Na沉积行为。降低Na2S的分解能垒可以极大地促进活性材料的利用,减少非活性Na2S的形成,从而达到较长的循环寿命。因此,系统地计算了Na2S在不同基底和N掺杂石墨烯(NC)上的分解能垒。图1a显示,与MnN4/C(1.54 eV)、ZnN4/C(1.84 eV)、NiN4/C(1.93 eV)、NC(2.00 eV)、CuN4/C(2.02 eV)、FeN4/C(1.87 eV)和CoN4/C(1.85 eV)相比,YN4/C在充电过程中对Na2S具有最小的分解势垒(1.52 eV),表明其能够促进Na2S的催化分解。
此外,Na2S6与YN4/C之间的结合能为−2.30 eV(图1b),明显强于Na2S6在其他基底上的结合能,这有利于硫还原反应并抑制多硫化物穿梭。图1c显示,YN4/C的电子离域比NC的要强烈得多。在YN4/C中,适度的电子离域可以降低电荷转移电阻,有利于均匀的金属沉积。
图1e的模拟钠沉积过程显示,在NC上,Na簇早在300ps就已经形成,而在YN4/C上即使在500ps也不会产生(图1d)。图1f显示了高度配位的Na原子数随时间的变化。图1g,h的电荷密度差分析显示,与NC相比,由YN4/C区组成的区域Na+与YN4/C之间的相互作用更强,说明Na+在这些活性位点上被有效吸收,协同促进了Na+的存储性能。图1i显示,Na从石墨烯晶格向Y中心的迁移以放热为主,说明这是一个自发过程。
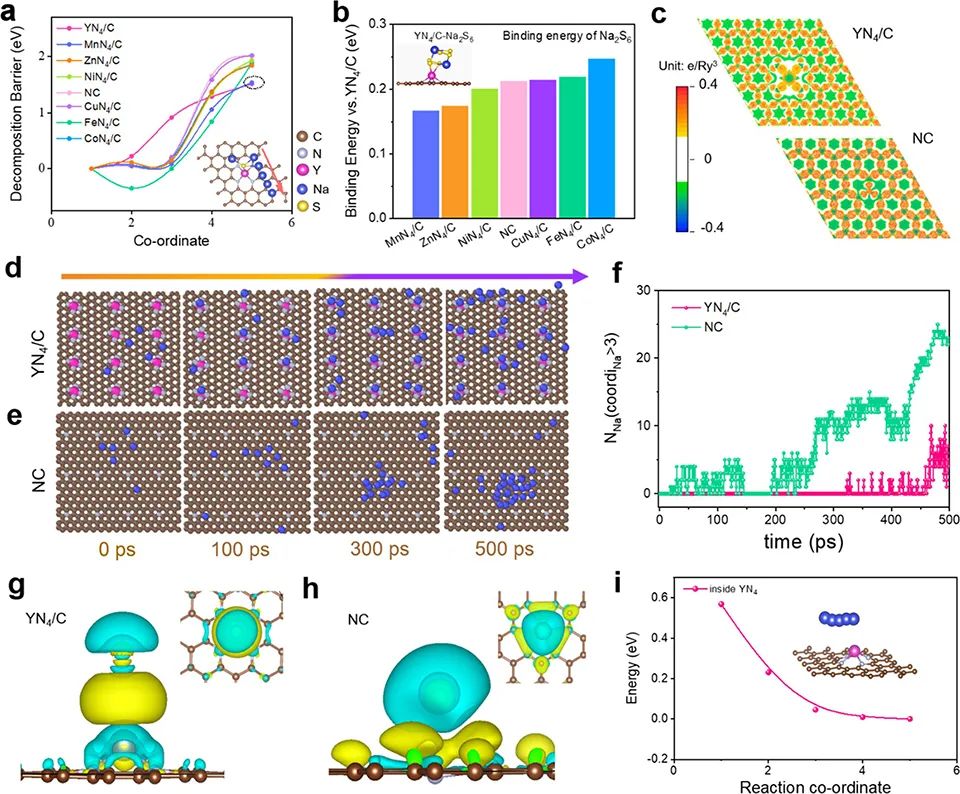
图 1、(a)Na2S在YN4/C、MnN4/C、ZnN4/C、NiN4/C、NC、CuN4/C、FeN4/C、CoN4/C等不同底物上的分解势垒。(b)相对于YN4/C,Na2S6在MnN4/C、ZnN4/C、NiN4/C、NC、CuN4/C、FeN4/C、CoN4/C上的吸附能。(c)YN4/C和NC的电子局域函数图。YN4/C(d)和NC(e)电极在特定模拟时间下的分子动力学模拟快照。(f)有3个以上Na配位的Na原子数随时间的变化。Na在YN4/C(g)和NC(h)电极上的吸附电荷密度差的俯视图和侧视图。(i)Na从石墨烯晶格迁移到YN4基团的扩散能垒。
从Y SAs/NC的TEM(图2a)可以看出,合成后的Y SAs/NC结构很好地保留了Y(acac)3/ZIF-8前驱体的初始菱形十二面体形态和尺寸分布。经像差校正的高角度环形暗场扫描透射电镜(AC-HAADF-STEM)图像(图2b)显示,存在高密度的单个亮点 (图2c),证明了Y在骨架上的原子级分散。能量色散X射线能谱图(EDS)显示,Y、C和N在整个十二面体中均匀分布(图2d)。
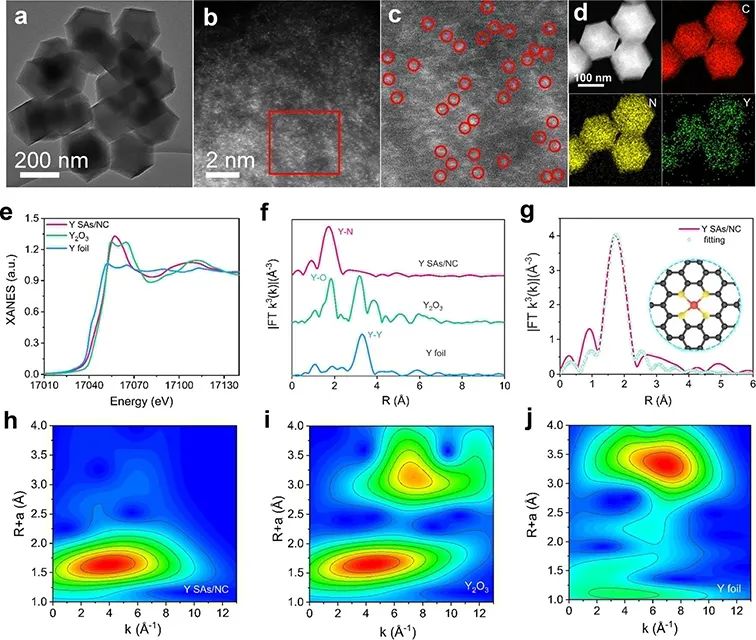
图 2、Y SAs/NC的TEM图像(a)和AC-HAADF-STEM图像(b)。(c)放大AC-HAADF-STEM图像。(d)Y SAs/NC的EDS元素映射。(e)不同样品的Y K边X射线吸收近边结构(XANES)光谱和(f)傅里叶变换(FT)k3加权扩展X射线吸收精细结构(EXAFS)光谱。(g)Y SAs/NC的EXAFS拟合图。(h-j)Y SAs/NC、Y2O3和Y箔的小波变换-EXAFS(WT-EXAFS)图。
进行了软X射线吸收近边结构(XANES)测量,以确定Y SAs/NC中N和C的电子结构。Y K边XANES谱的阈值能量位于Y箔和Y2O3的阈值能量之间(图2e),说明Y在Y SAs/NC中的平均氧化态约为2.31。图2f中Y SAs/NC的FT-EXAFS分析显示,在1.71 Å附近有一个主峰,这是由于第一壳层Y-N路径的散射。
此外,与Y箔相比,Y SAs/NC中未检测到主峰在3.28 Å左右的Y-Y键,说明Y原子分散在碳基体上,具有Y-N配位。 Y SAs/NC的配位构型为一个完美平面YN4配位,如图2g所示。
此外,在k和R空间中进行了高分辨率的小波变换EXAFS(WT-EXAFS),以识别Y SAs/NC中Y物种的原子分散状态。如图2h–j所示,Y SAs/NC的WT计数曲线在3.9°-1左右出现最大信号,位于Y箔(Y–Y,6.8°-1)和Y2O3(Y–O,4.3°-1)之间,表明Y–N键在Y SAs/NC中占主导地位。
图3a 显示,在1.0 mA cm-2@1.0 mAh cm-2下,Y SAs/NC能够循环400次,具有稳定的库仑效率(CE),超过99%。而NC和裸Cu的CE波动较大,这主要是由于Na金属负极表面沉积了Na枝晶或死Na。图3b显示,Y SAs/NC-Na在三个电极中极化最小。在1.0 mA cm-2@1.0 mAh cm-2下,Y SAs/NC的成核过电位低至22.4 mV,低于NC(32.9 mV)和Cu(48.4 mV)电极的成核过电位,表明Y SAs/NC具有良好的亲钠性,能够实现均匀的Na沉积动力学。
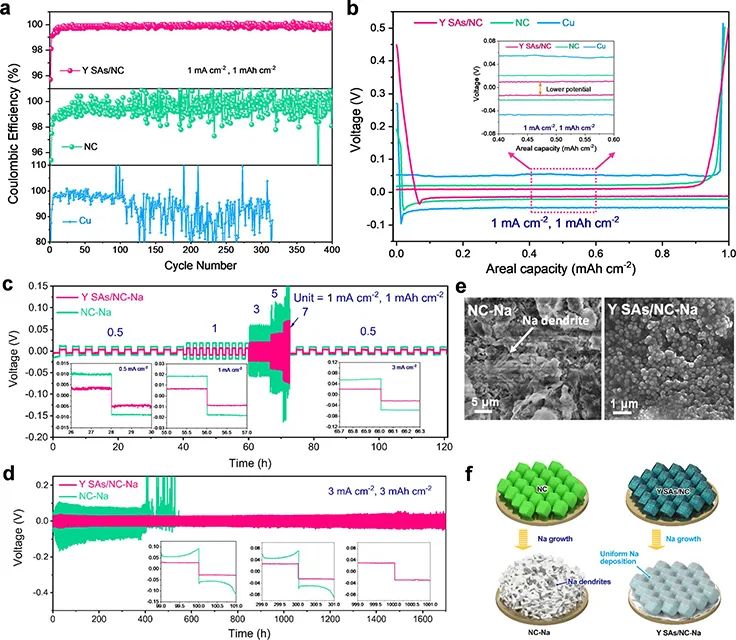
图 3、(a)1.0 mA cm-2@1.0 mAh cm-2下,不同电极的库仑效率和(b)电压分布。(c)Y SAs/NC-Na|Y SAs/NC-Na和NC-Na|NC-Na对称电池倍率性能。(d)Y SAs/NC-Na和NC-Na对称电池长循环稳定性。(e)循环后的NC-Na和Y SAs/NC-Na负极SEM图像。(f)Na在NC和Y SAs/NC电极上的电镀行为示意图。
图3c显示,Y SAs/NC-Na电极表现出稳定的倍率性能和较低的极化,而在5 mA cm-2时,NC-Na对称电池产生了巨大的电压波动。图3d显示,在3 mA cm-2@3 mAh cm-2下,Y SAs/NC-Na电极能够稳定循环1500 h,而NC-Na电极只能循环小于550 h(图3d)。
图3e显示,在3 mA cm-2@3 mAh cm-2循环后,NC-Na电极形成了明显的“空腔”和Na枝晶。而Y SAs/NC没有观察到Na枝晶。图3f显示,金属Na以均匀颗粒的形式沉积在Y SAs/NC-Na复合材料上,这得益于YN4位点的调节作用。
图4a的循环伏安(CV)曲线显示,Y SAs/NC-S正极有两个清晰的阴极峰,对应于固态硫还原为可溶的长链NaPS(Na2Sx, 4
Y SAs/NC-S的阳极和第二个阴极峰之间的电压差为0.26 V,明显低于NC-S的0.34 V极化,表明YN4基团抑制了电化学极化,改善了转化动力学。
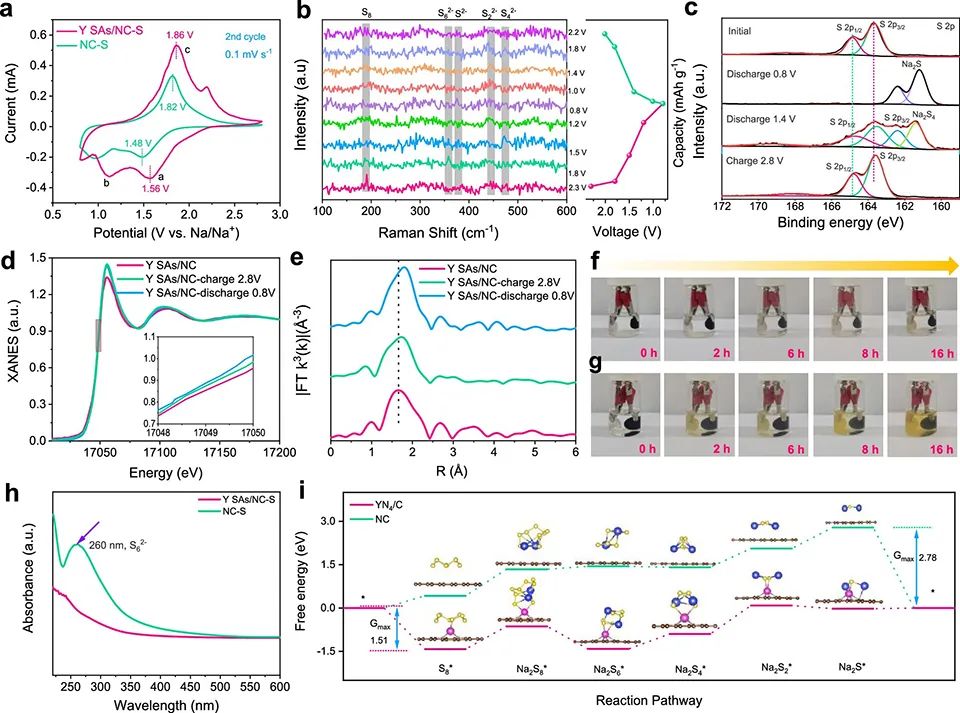
图 4、(a) 在0.1 mV-1 s-1下,Y SAs/NC-S和NC-S正极的CV曲线。(b)由Y SAs/NC-S组成的室温Na-S电池原位拉曼光谱。(c)Y SAs/NC-S正极在不同充放电状态下的非原位XPS光谱。(d)在0.1 A g-1下,Y SAs/NC-S在原始状态和不同充放电状态下的Y K边XANES光谱。(e)不同状态下对应的Y K边傅里叶变换(FT)k3加权EXAFS光谱(R空间)。从初始状态到不同放电状态,Y SAs/NC-S(f)和NC-S的颜色变化。(h)16 h放电循环后Y SAs/NC-S和NC-S的紫外-可见(UV-vis)光谱。(i)计算Y SAs/NC和NC上S物种逐步还原的吉布斯自由能。
图4b的原位拉曼光谱显示,当电池放电至1.8 V时,190.7 cm-1处的S拉伸振动带消失,并出现另一个峰(≈358 cm-1),该峰值可对应Na2S6。在随后的放电过程中,Na2S4在约444 cm-1处进一步转化为Na2S2。放电结束时,Na2S在≈375 cm-1处形成。
随后,Na2S氧化为Na2Sx(1.8 V),然后Na2Sx依次转化为长链多硫化钠(2.2 V),最后在Y SAs/NC催化下可逆转化为S。 图4c的XPS显示,在放电至1.4 V时,检测到位于161.4和162.4 eV的双峰,对应Na2S4。在163.6和164.8 eV处产生了新的峰,这可以归因于Na2S。这表明Y SAs/NC-S具有优越的反应动力学。
在Y K边XANES光谱中(图4d),由于氧化Y态的还原,Y SAs/NC-S中Y元素的K吸收边在放电状态下向能量较初始Y SAs/NC低的方向移动。在随后的充电过程中,吸收边逐渐移回到更高的能量,说明络合物转移回了原始状态(图4e)。这证实Y SAs/NC-S表面上的键合NaPS失去电子并转化为硫,因此,Y位的还原状态也被可逆地氧化。
图4f显示,在Y SAs/NC-S电池中,电解液在放电16小时后仍然是无色的,而在NC-S电池中,电解液在放电2小时后变为微淡黄色(图4g)。这归因于YN4位点对可溶性多硫化钠的吸附。UV-vis光谱(图4h)显示,Y SAs/NC中Na2S6的强度下降(峰在260 nm处),证实了NaPS与Y单原子之间有很强的化学亲和力。
图4i计算的基于S8和Na可逆生成Na2S的整个反应能量路径显示,在放电过程中,首先是S8吸附,然后S8与两个Na+双还原生成Na2S8,随后Na2S8进一步还原,形成Na2S6、Na2S4、Na2S2三个NaPS中间产物,最终生成Na2S。对于NC,由于吸附相当弱,没有发现占主导地位的吸附物质,而Na2S*是决速组分。
对于YN4/C, Na2S6*和Na2S2*分别为主要吸附组分和决速组分。NC和YN4/C的Gmax分别为2.79和1.51 eV,证实Y SAs/NC上的硫还原过程比NC上的更容易。 将Y SAs/NC Na负极和Y SAs/NC-S正极配对组装Na–S全电池(图5a)。图5b显示,Y SAs/NC-S||Y SAs/NC-Na表现出优越的倍率性能。此外,在5 A g-1下,经过1000次循环后,其容量达到510 mAh g-1,高于Y SAs/NC-S||Na的容量(图5c)。
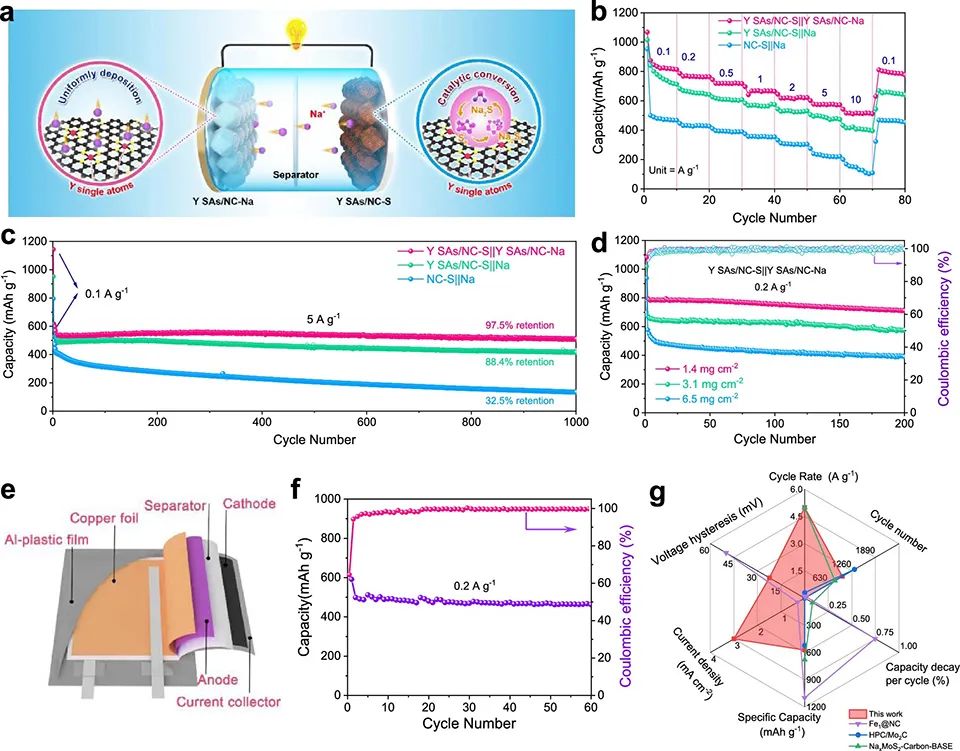
图 5、(a)Y SAs/NC-S||Y SAs/NC-Na全电池示意图。(b)Y SAs/NC-S||Y SAs/NC-Na, Y SAs/NC-S||Na和NC-S||Na全电池倍率性能。(c)在5 A g-1下,不同全电池的长循环稳定性。(d)Y SAs/NC-S||Y SAs/NC-Na全电池与3D打印Y SAs/NC-S||Y SAs/NC-Na全电池循环性能的比较。(e)具有Y SAs/NC-S正极的柔性Na-S软包电池示意图。(f)Na-S软包电池在0.2 A g-1下的循环性能。(g)Y SAs/NC-S||Y SAs/NC Na电池与一些最先进的Na-S体系的比较。
此外,Y SAs/NC-S||Y SAs/NC Na全电池在0.2 A g–1下可提供787 mAh g–1的容量(图5d)。载量为3.1和6.5 mg cm–2的3D打印全电池在0.2 A g–1下第五次循环容量分别为656和522 mAh g–1。即使在0.2 A g–1下,6.5 mg cm–2硫载量的全电池进行200次循环后,仍能保持近100%的库伦效率,且每个循环的衰减率低,为0.33%(图5d)。
此外,还组装了一个Y SAs/NC-S软包电池(图5e)。将软包电池载量增加至∼80 mg(对应∼2.3 mg cm–2)。图5f显示,在0.2 A g–1下,组装后的电池容量高达500 mAh g–1。图5g显示,Y SAs/NC-S||Y SAs/NC Na性能优于一些最先进的Na–S体系。
5
总结与展望
本工作设计了基于Y SAs/NC-S正极和Y SAs/NC-Na负极的新型Na-S全电池。理论预测表明,YN4/C具有优化的电荷结构和可调的电子局域化特征,可以促进均匀的Na沉积,防止枝晶生长,加速NaPS转换,降低Na2S分解能垒。Y SAs/NC作为同时调节S正极和Na负极的Janus功能宿主,对促进S氧化还原反应动力学和抑制多硫化物穿梭过程具有协同作用,抑制了Na枝晶生长。
构建的Y SAs/NC-S||Y SAs/NC-Na全电池在0.1 A g-1下具有822 mAh g-1的容量。此外,采用Y SAs/NC-S正极的软包电池在0.2 A g-1下具有500 mAh g-1的高面积容量,证明了其在柔性Na-S电池中的实际应用前景。本工作为制备高性能Na-S全电池提供一种实用的策略。
审核编辑:刘清