直接硼氢化钠/双氧水燃料电池研究
摘要:采用Pt/C作为催化剂成功地组装了直接硼氢化钠/双氧水燃料电池,并考察了不同操作温度、溶液流速和浓度 条件下电池的放电性能。 60℃时电池的最大比功率可以达到130 mW/cm2,在40℃时0.1 A/cm2放电条件下电池电 压约为0.7 V,性能明显优于相同条件下直接甲醇燃料电池。同时研究了不同厚度电解质膜对电池性能的影响,采用 Nafion117膜(厚度175 mm)的电池开路电压比Nafion112(厚度 50 mm)高约180 mV,但Nafion112在高电流密度 放电时表现出了更好的性能。因此,膜厚度不仅影响电池的内阻而且还会影响反应物的相互渗透。此外,还测试了短时 间恒电流放电,电池性能未出现下降,而且放电后催化层和膜仍然保持紧密结合。
使用氢气的质子交换膜燃料电池近些年来获得了广泛的 关注,尤其是在电动汽车等大型牵引动力源方面研究取得了 很多进展。但由于氢气储存困难,便携性差等不足限制了其在 小型可移动电源方面的应用,人们一直试图寻找替代氢气的 液体燃料,其中以甲醇作为燃料的直接甲醇燃料电池(DM- FC)被认为在此领域大有作为。然而甲醇氧化速度慢,甲醇渗 透等问题一直没有彻底解决,使得DMFC商业化过程中遇到了很多阻力[1]。 硼氢化物同样作为一种液体燃料,具有便于储存、运输等 特点,更为重要的是它的活性要远高于醇类燃料,而且如果采 用双氧水作为氧化剂,整个反应将不依赖空气,有望在一些特 殊领域例如水下和航天方面的便携式电源获得应用[2-4]。 硼氢化钠作为反应燃料的反应方程式如下[5-6]:
通过理论计算可以得出硼氢化钠燃料电池的开路电压和 比能量分别为1.64 V和9 295 Wh/kg比甲醇的开路电压和比 能量分别高0.4 V和 3 200 Wh/kg[6]。
对于阴极用氧气作为燃料的硼氢化钠燃料电池,仍然存 在与甲醇燃料电池一样的阴极气体储存携带不便的问题,为 此,研究者提出了以H2O2代替O2的 NaBH4/H2O2燃料电 池[7-8]。如果阴极H2O2为碱性,则阴极反应为:
近几年来,很多研究者在催化剂材料选择方面作了大量 的研究。为了解决阳极催化剂活性低的问题,Gyenge等人提 出了胶体Os、Au、Pt和Os、 Au、Pt基的合金催化剂,实验发 现,Pt-Ni和Pt-Ir合金催化剂有最好的活性[9-11];Chatenet等人 研究了Au和Ag块材和纳米颗粒分散在碳载体的催化剂,实 验发现纳米分散在碳上的Au催化剂具有最好的活性[12]; Feng等人利用MnO2作为阴极催化剂发现既有较好的氧还原 活性又可以减少硼氢化钠渗透所导致的阴极极化[13]。此外,针 对硼氢化钠的渗透,研究人员又从膜和催化剂两方面进行了 研究,Lakeman测试了十二种不同的质子交换膜[14],Shukla等 人报道了利用Nafion961膜制作的电池在减少硼氢化钠渗透 方面比传统的使用 Nafion117有了较大的提高[15];然而,系统 研究操作条件对硼氢化钠/双氧水电池性能影响的文章较 少。
本文通过单电池评价装置测试了NaBH4/H2O2燃料电池 的极化曲线,系统研究了不同工作温度、浓度、流速对电池性 能的影响,而且比较了不同厚度 Nafion膜对电池性能的影 响。同时考察了电池的短期运行稳定性。
1实验内容
1.1 Nafion膜
Nafion膜在使用前进行预处理,以去除膜中的有机物和 金属离子。具体做法如下:将一定尺寸的Nafion膜依次在3% (质量分数)的H2O2、去离子水、0.5 mol/L的H2SO4、去离子水 中处理,每次处理时间为1 h,处理温度为80℃。处理后的 Nafion膜保存在去离子水中备用。
1.2电极的制备
催化剂采用Johnson Matthey公司的40%的Pt/C。具体的 制备工艺如下:取少量Pt/C催化剂放入乙醇中超声分散成 “油墨”状,然后滴加Nafion溶液,其中催化剂与Nafion干重 质量比为9︰1,超声后均匀涂在扩散层碳纸上即得到电极。 实验中Pt载量为 1.4 mg/cm2。实验所用的阴极与阳极电极均 为相同的Pt/C电极,电极活性面积2.5 cm×2.5 cm。
1.3膜电极三合一的制备
将阳极和阴极催化剂刷涂的一面朝向Nafion膜,在1.5 MPa,140℃下热压3 min,制成膜电极三合一(MEA)。
1.4溶液的配制
阳极硼氢化钠溶液采用NaBH4含量为10%,其中含有 5%NaOH,5%NH·3H2O;阴极双氧水采用H2O2含量为10%,其 中含有 5%H3PO4。对于不同浓度的NaBH4和H2O2其中各组 分的比例与上面的配比一致。
1.5电池性能的测试
将上述制好的膜电极三合一组装单电池。集流板采用高 纯石墨制备,流场为单通道蛇形。两侧用不锈钢板通过螺栓夹 紧,并置入NaBH4/H2O2燃料电池的评价装置中。
不同浓度的NaBH4和H2O2分别通过恒流泵以设定的流 速进入电池阳极和阴极,反应后的废液分别流进各自的废液 瓶,不再循环使用,以保证实验过程中反应物质的浓度保持不 变。电池工作时通过热电偶和温度控制装置将其保持在设定 的温度。电池的极化曲线测量采用日本菊水电子的PLZ4电 子负载进行自动控制,在每个电流点进行恒电流放电,直到电 池电压不再发生明显的变化,再进行下一个点的测量,终止电 压为0.2 V。
2结果与讨论
2.1温度对直接硼氢化钠燃料电池性能的影响 图1是利用自制的电极和Nafion112膜组装的电池在不 同温度下的工作性能曲线。其阳极NaBH4和阴极H2O2的浓 度均为10%,电池的阳极阴极流速分别为0.8 mL/min,3.2 mL/min。从图中可以看出,随着温度的升高,电池的最大比功 率有较大的提高,开路电压也同时有所提高。在40℃时电池 的开路电压为1.39 V,最大比功率为74 mW/cm2;当温度升高 至60℃,开路电压和最大比功率分别升高至1.42 V和99 mW/cm2;继续升温到80℃会使电池开路电压比40℃时提高 了60 mV,最大比功率可达 117 mW/cm2。
电池温度的升高,一方面增加了Nafion膜中Na+的传递 速度,降低了电池的整体内阻,从而提高电池放电能力;另外 一方面,温度的升高还将提高 NaBH4电化学氧化和H2O2电 化学还原反应速度,降低了反应的过电位,使得电池的开路电 压获得提高[16]。
2.2不同流速对单电池性能的影响
图2和图3分别是Nafion112膜组装的电池在不同流速下的电池性能曲线,实验中的温度固定在60℃,阴极阳极液 体浓度均为10%。在图2中阳极流速固定在0.8 mL/min,可以 看出,随着阴极流速的增加,电池的开路电压从阴极流速为2 mL/min时的1.41 V增加至5.2 mL /min时的1.43 V,电池最 大比功率相应的从83 mW/cm2提高到了93 mW/cm2。在图3 中阴极流速固定在3.2 mL/min,可以看出随着阳极流速由 0.4mL/min升高至0.8 mL/min时,电池的最大比功率提高了 14 mW/cm2,开路电压升高了10 mV,然而当阳极流速升高至 1.6 mL/min时,电池的开路电压比0.4 mL/min时还减少了10 mV,电池的最大比功率比1.6 mL/min时减少了3 mW。
当阳极流速一定时,随着阴极流速的增加,阴极传质速率 加快,流速的加快同时也会减少催化层孔洞的堵塞和产物的 积累,从而使电池的性能提升[16];当阴极流速一定时,随着阳 极流速增加,开始时电池的开路电压和最大功率会相应的增 加,但随着流速的加大,性能则有所下降。这可能是由于高流 速下 NaBH4的渗透增加导致的阴极性能降低而造成的电池 性能整体下降。渗透到阴极的NaBH4会使阴极产生混合电位 从而降低了电池的开路电压。因此,在实际应用的时候,阳极 NaBH4的流速应该选择在一个较佳的值。
2.3不同原料浓度对单电池性能的影响
图4和图5分别是电池采用不同浓度H2O2和NaBH4时 电池的性能曲线,实验中温度固定在60℃,阳极流速0.8 mL/min,阴极流速为 3.2 mL/min。从图4中可以看出当阳极浓 度固定为10%NaBH4时,随着阴极H2O2的浓度增加,电池的开路电压及比功率均有大幅提高。阴极 H2O2的浓度为5%时, 电池的开路电压仅为1.35 V,最大比功率为88 mW/cm2;当阴 极改为10%的H2O2时,电池开路电压升高为 1.45 V,最大比 功率增加到96 mW/cm2;继续增加阴极H2O2的浓度至20% 时,电池的开路电压升高至1.52 V,最大比功率超过了 130 mW/cm2。图5为固定阴极H2O2的浓度为10%时,改变阳极 NaBH4的浓度,当阳极浓度由5%升高至10%时,电池的开路 电压和最大比功率分别提高了5 mV和9 mW;当NaBH4的 浓度升高至20%时,电池的开路电压升高至1.45 V,然而其最 大比功率下降至 80 mW/cm2,而且电池在高电流密度下的放 电明显下降,200 mA/cm2放电时电池电压下降至0.2 V,而利 用5%和10%的NaBH4 时该值分别为0.455 V和0.502 V。
从上面的数据可以看出当阳极浓度固定时,随着阴极 H2O2浓度的增加,阴极的传质速率加快,进而使反应速率加 快,从而电池的开路电压和最大比功率提高;当阴极浓度固定 时,阳极NaBH4的浓度增加,一方面会提高阳极的传质速率, 但同时也会增大浓差极化和NaBH4的水解,导致阳极的 NaBH4 渗透较多进而增加了阴极极化,因此会出现实验中浓 度超过某一值时的电池性能下降的现象[16]。
2.4不同Nafion膜对于直接硼氢化钠燃料电池性 能的影响
图6是在相同的工作条件下(60℃;阳极NaBH4浓度为 10%,流速0.8 mL/min;阴极H2O2浓度为10%,流速3.2 mL/min)分别用Nafion112(厚度50μm)和Nafion117(厚度 175μm)制作的电池性能比较。从图中可以看出,随着Nafion膜厚度的增加,电池的开路电压升高,这主要是由于低电流密 度下NaBH4渗透对电池性能起主要作用,随着膜厚度的增加 NaBH4渗透逐渐减少,阴极极化减少,从而使阴极电极电位升 高,导致电池开路电压升高。然而随着放电电流密度的增加, NaBH4渗透产生的阴极极化不再起主要作用,而Nafion膜本 身的膜电导成为控制电池性能的主要因素,Nafion膜越薄则 膜电阻也越小,从而电池性能越好。
2.5稳定性实验
对于利用Nafion112膜制作的电池还进行耐久性试验, 放电条件是分别在50、100、150 mA/cm2的电流密度下放电 30 min,得到的放电曲线如图7所示。可以看出,整个电池在 短期实验中是有部分时间不稳定的,并且在不同电流密度放 电的情况下不稳定的情况都有发生,因此这种现象是与放电 无关的,而是电池自身的原因。分析现象可能是因为阳极 NaBH4水解产生氢气或阴极H2O2分解产生氧气造成,由于放 电的电子负载记录时间间隔为1 s,有可能在很短的时间内生 成的气体将反应的液体隔断而造成瞬时电阻增大导致性能的 瞬间衰减,不过电池电压会很快恢复到原来的性能。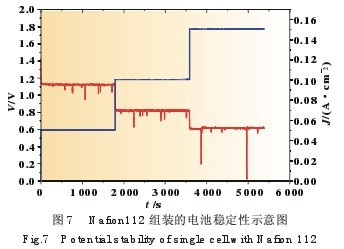
所以说,在直接硼氢化钠燃料电池研究过程中,如何找到 使阳极发生8电子转化反应、减少NaBH4水解产生氢气的阳 极催化剂,并且研发可以催化还原 H2O2但不使其分解的阴极 催化剂是下一步研究的重要方向。
图8为利用Nafion112制作的电池在60℃下相同浓度 (阳极NaBH4:10%,阴极H2O2:10%)、流速(阳极 NaBH4: 0.8 mL/min,阴极H2O2:3.2 mL/min)下几次不同实验的对比,
时间的前后间隔约为一个星期。从中可以看出,电池的性能基 本重复没有明显衰减。
图9为经过测试后的膜电极集合体截面的SEM图。可以 明显看出经过测试后的MEA的三相反应界面仍然保持完 整,Nafion膜和催化层粘结牢靠并未出现剥离现象。
3结论
本文采用Nafion112成功地组装了直接硼氢化钠/双氧 水燃料电池,重点考察查了工作温度、燃料浓度、流速等操作 条件对电池性能的影响;并且比较了不同厚度Nafion膜组装 的电池性能。实验结果表明:电池的开路电压随温度的升高而 升高;阳极流速和浓度对电池的开路电压影响不大,当流速超 过某一特定值时电池的性能开始下降,当阳极NaBH4浓度过 高时,会使电池的最大比功率和开路电压大为降低,这主要是 由于高浓度NaBH4渗透和水解造成的;电池的开路电压及最 大比功率均随阴极的流速及浓度的提高而增大。对于不同 Nafion膜组成的电池,可以看出Nafion117比 Nafion112开路 电压高,而高电流密度放电下Nafion112性能较好,这说明膜 电导和NaBH4渗透同时影响电池的性能;短期的恒电流放电 和SEM结果表明电池的稳定性比较理想。
参考文献:
[1]刘建国,衣宝廉,魏昭彬.直接甲醇燃料电池的原理、进展和主要技 术问题[J].电源技术,2001,25(5):363-366.
[2]DOO H J,CHANG H L,CHANG H K,et
al.Performance of a direct
methanol polymer electrolyte
fuel cell[J].J Power Sources,
1998,71(1/2):169-173.
[3]PONCE DE LEON C,WALCH F C,
PLETCHER D,et al.Direct
borohydride fuel cells[J].J
Power Sources,2006,155(2):172-181.
[4]JUNG H W.A comparison of sodium
borohydride as a fuel for
proton exchange membrane fuel
cells and for direct borohydride fuel
cell[J].J Power Sources,2006,155(2):329-339.
[5]LI Z P,LIU B H,ARAI K,et al.
Development of the direct borohy-
dride fuel cell[J].J Alloys and
Compounds,2005,404-406:648-652.
[6]UMIT B DEMIRC I.Direct borohydride
fuel cell:Main issues met
by the membrane-electrodes-assembly
and potential solutions[J].J
Power Sources,2007,172(2):676-687.
[7]GEORGE H M,NIE L,JOSEPH M,et
al.Direct NaBH4/H2O2 fuel
cells[J].J Power Sources,
2007,165(2):509-516.
[8]CHOUDHURY N A,RAMAN R K,
SAMPATH S,et al.An alka-
line direct borohydride fuel
cell with hydrogen peroxide as
oxidant [J].J Power Sources,2005,143(1/2):1-8.
[9]ATWAN M H,NORTHWOOD D O,GYENGE
E.Evaluation of colloidal Os
and Os-alloys(Os-Sn,Os-Mo and Os-V)for
electro- catalysis of methanol and
borohydride oxidation[J].Int J Hydrogen
Energy,2005,30(12):1323-1331.
[10]ATWAN M H,MACDONALD C L B
,NORTHWOOD D O,et al. Colloidal
Au and Au-alloy catalysts for direct
borohydride fuelcells:Electrocatalysis
and fuel cell performance[J].J Power
Sources,2006,158(1):36-44.
[11]GYENGE E,ATWAN M H,NORTHWOOD
D O.Electrocatalysis of borohydride
oxidation on colloidal Pt and Pt-alloys
(Pt-Ir,Pt-Ni, and Pt-Au)and application
for direct borohydride fuel cell anodes
[J].J Electrochem Soc,2006,153(1):A 150-A 158.
[12]CHATENET M,MICOUD F,ROCHE I,
et al.Kinetics of sodium borohydride
direct oxidation and oxygen reduction
in sodium hy- droxide electrolyte-Part
I.BH4-electro-oxidation on Au and Ag
catalysts[J].Electrochim Acta,
2006,51(25):5459-5467.
[13]FENG R X,DONG H,WANG Y D,et
al.A simple and high effi-
cient direct borohydride fuel
cell with MnO2-catalyzed cathode[J].
Electrochem Commun,2005,7(4):449-452.
[14]LAKEMAN J B,ROSE A,POINTON
K D,et al.The direct boro-
hydride fuel cell for UUV
propulsion power[J].J Power Sources,
2006,162(2):765-772.
[15]RAMAN R K,PRASHANT S K,SHUKLA
A K.A 28-W portable direct borohydride
hydrogen peroxide fuel-cell stack[J].
J Power Sources,2006,162(2):1073-1076.
[16]CHENG H,SCOTT K.Influence of
operation conditions on direct
borohydride fuel cell performance[J].
J Power Sources,2006,160 (1):407-412.